
Tumores embrionarios del SNC
96
Tumores Embrionarios MC, MSc. Sandro Casavilca Zambrano Profesor de la Facultad de Medicina de San Fernando UNMSM Patólogo Oncólogo. Jefe de la Unidad Funcional de Patología Quirúrgica y Banco de Tejidos Tumorales Instituto Nacional de Enfermedades Neoplásicas
-
Upload
sandro-casavilca-zambrano -
Category
Health & Medicine
-
view
84 -
download
2
Transcript of Tumores embrionarios del SNC

Tumores Embrionarios
MC, MSc. Sandro Casavilca ZambranoProfesor de la Facultad de Medicina de San Fernando UNMSMPatólogo Oncólogo. Jefe de la Unidad Funcional de Patología Quirúrgica y Banco de Tejidos TumoralesInstituto Nacional de Enfermedades Neoplásicas

Objetivo
EpidemiologíaCausasDiagnóstico y clasificación de los tumores embrionarios del SNCComentarios finales/Próximos pasos

1. Epidemiología:



Tumores del Sistema Nervioso Central
Meninges (C70) Encéfalo (C71)Médula espinal, nervios craneales y otras partes
del SNC (C72)
Astrocitoma pilocítico 0 43 2Astrocitoma difuso 0 126 1 Astrocitoma anaplásico 0 100 0Glioblastoma 0 360 3Oligodendroglioma 0 151 0Oligodendroglioma anaplásico 0 66 1Oligodendroglioma - oligoastrocitoma anaplásico 0 96 0
Meduloblastoma 0 174 0T. neuroectod.primitivo central 0 16 3
Tumor teratoide rabdoide atípico 0 9 0
Ependimoma 0 54 25Meningioma 248 0 2Tumor de células germinales 0 8 0Linfoma primario del sistema nervioso central 1 30 3
Schwannoma 0 7 8Schwannoma maligno 0 0 1Ganglioma 0 3 0
Meninges (C70) Encéfalo (C71)Médula espinal, nervios
craneales y otras partes del SNC (C72)
Total de número de casos 249 1243 49Porcentaje 16.2 80.7 3.2
INEN Registro de Tumores SNC
Tumores del Sistema Nervioso Central
(2005 - 2014)

24%
16%
11%10%
8%
6%
6%
5%
4%
3%2% 1%
Tumores del Sistema Nervioso Central - INEN(2005 - 2014)
Glioblastoma
Meningioma maligno
Meduloblastoma
Oligodendroglioma
Astrocitoma difuso
Astrocitoma anaplásico
Oligodendroglioma - oligoastrocitoma anaplásico
Ependimoma
Oligodendroglioma anaplásico
Astrocitoma pilocítico
Linfoma primario del sistema nervioso central
T. neuroectod.primitivo central
Schwannoma maligno
Tumor teratoide rabdoide atípico
Tumor de células germinales
Ganglioma anaplásico
Schwannoma

2. Tumores del SNC CausasLos principales trastornos genéticos son producidos por las radiaciones ionizantesSíndromes hereditarios(como neurofibromatosis, Esclerosis tuberosa , Síndrome de Li-Fraumeni, Von Hippel-Lindau, Turcot y Gorlin)

Síndromes Hereditarios
Síndrome de Gorlin (NBCCS):Gen PTCH en 9q22.3 (o Gen SUFU en 10q24.32)Infantes, meduloblastoma desmoplásico/nodular/SHH
Turcot tipo 2:Gen APC en 5q21FAP, cáncer de colon y meduloblastomas
Síndrome Li Fraumeni:Gen TP53 en 17p13Meduloblastomas, PNETs y CPCs
Síndrome de predisposición Rabdoide:SMARCB1 (INI1) Gen en 22q11.2ATRTs y MRTs en niños

3. Diagnóstico de Neoplasias Neuronales / Embrionarias
Ganglio = Neuronas maduras grandesNeurocito= Neuronas maduras pequeñasNeuroblasto = Neuronas primitivas pequeñasParaganglio = Célula autonomica neuroendocrinaEstesio = Célula neuronal olfatoriaPineo = Célula neuronal pinealRetino = Célula neuronal retinal
Clasificación OMS 2016Libro azul OMS 2007 Libro azul OMS 2016

¿Que ha cambiado?
SOMOS LOS MISMOS PERO MEJORADOS …........

Tumores EmbrionariosOMS 2007
MeduloepiteliomaEpendimoblastomaMeduloblastomaTumor neuroectodermal primitivo (PNET)Tumor Teratoide/Rabdoide atípico
OMS 2016
Meduloblastoma genéticamente definidoMeduloblastoma histológicamente definidoMeduloblastoma NOSTumor embrionario con rosetas de múltiples capas C19MC alteradoTumor embrionario con rosetas de múltiples capas NOSMeduloepiteliomaNeuroblastoma del SNCGanglioneuroblastoma del SNCTumor embrionario NOS del SNCTumor teratoide/rabdoide atípicoTumor embrionario del SNC con cambios rabdoides

MEDULOBLASTOMAS
Tumor embrionario anaplásico de células pequeñas del cerebeloNo relacionado con otros tumores embrionariosNo relacionado con el PNET periférico (sarcoma de Ewing)

MEDULOBLASTOMAS (cont)
TIENE UNA INCIDENCIA DE 0.5 POR 100000/año.SE LOCALIZA EN CEREBELO75% DE LOS MEDULOBLASTOMAS EN LA INFANCIA SE ORIGINAN EN EL VERMISEL DEFECTO CROMOSOMICO ESTA EN EL CROMOSOMA 17 EN LA MAYOR PARTE DE LOS CASOS.SON POSITIVOS PARA SINAPTOFISINAIndice proliferativo (Ki67); >20%

MEDULOBLASTOMAEPIDEMIOLOGIA
1° década de la vida(15-20 % de todos los tumores intracraneales infantiles y el más frecuente de los tumores cerebrales malignos en edad pediátrica) . Segundo pico entre los 20-30 añosPredominio masculino 2:1 con respecto a mujeres.10 a 35 % diseminación en el momento del diagnóstico.

MEDULOBLASTOMA
MACROSCOPIATumor infiltrante, de aspecto encefaloideoVermis cerebeloso, en ápex del techo del 4° ventrículo (niños), hemisferios cerebelosos (adultos)Gran tendencia a metastatizar en eje craneoespinal, por LCR, excepcionalmente fuera del SNC (huesos largos).
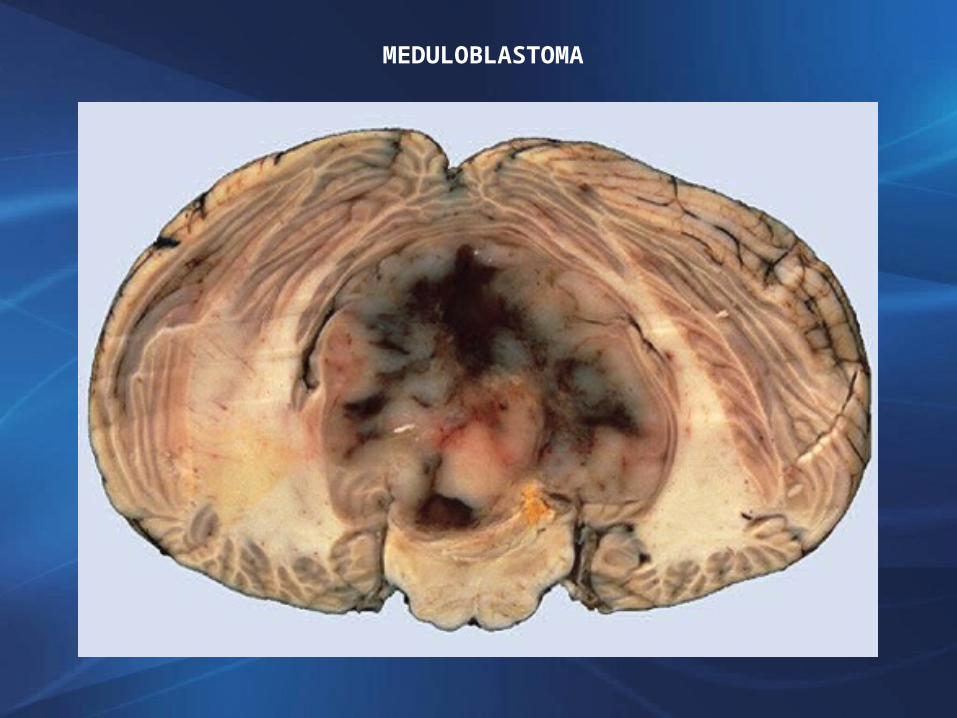
MEDULOBLASTOMA

EtiologíaGorlin: Tipo nodular / desmoplásico (raro)Turcot tipo 2, APC: Clásico (no nodular) (raro)
HistologíaClásico: ~ 70%Nodular / desmoplásico: ~ 15-20%Células grandes / anaplásicas: grado de anaplasia requerido y porcentaje de área de sección transversal tumoral poco clara, pero generalmente anaplasia severa y / o tipo de células grandes de más del 50% o más del área tumoral

Variantes Histológicas
MB Clásico
MB Desmoplásico/Con extensa nodularidad
MB de células grandes/ MB anaplásico



Meduloblastoma con Extensa Nodularidad
ICD-O 9471/3OMS GRADO IVArquitectura expansiva, lobular debido a que las zonas libres de reticulina llegan a ser inusualmente grandes. Se presenta en infantes y está íntimamente relacionado con el meduloblastoma desmoplasico/nodular


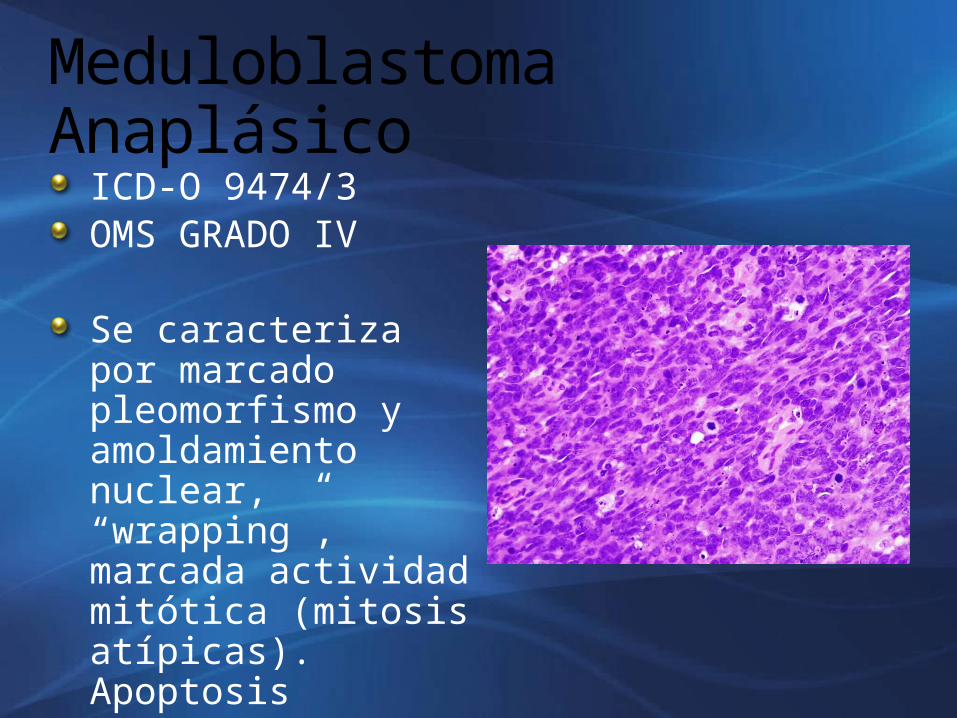
Meduloblastoma Anaplásico
ICD-O 9474/3OMS GRADO IV
Se caracteriza por marcado pleomorfismo y amoldamiento nuclear, “wrapping”, marcada actividad mitótica (mitosis atípicas). Apoptosis

CRITERIOS DE ANAPLASIAA. Incremento del tamaño nuclear B. Abundantes mitosisC. Numerosas apoptosisD. Pleomorfismo con nucleolos prominentes y
amoldamiento nuclear.Expresión de p53, c-myc y n-myc se asocian a mal pronóstico.
TRKC (receptores afines a la neurofilina) SE ASOCIA A BUEN PRONÓSTICO
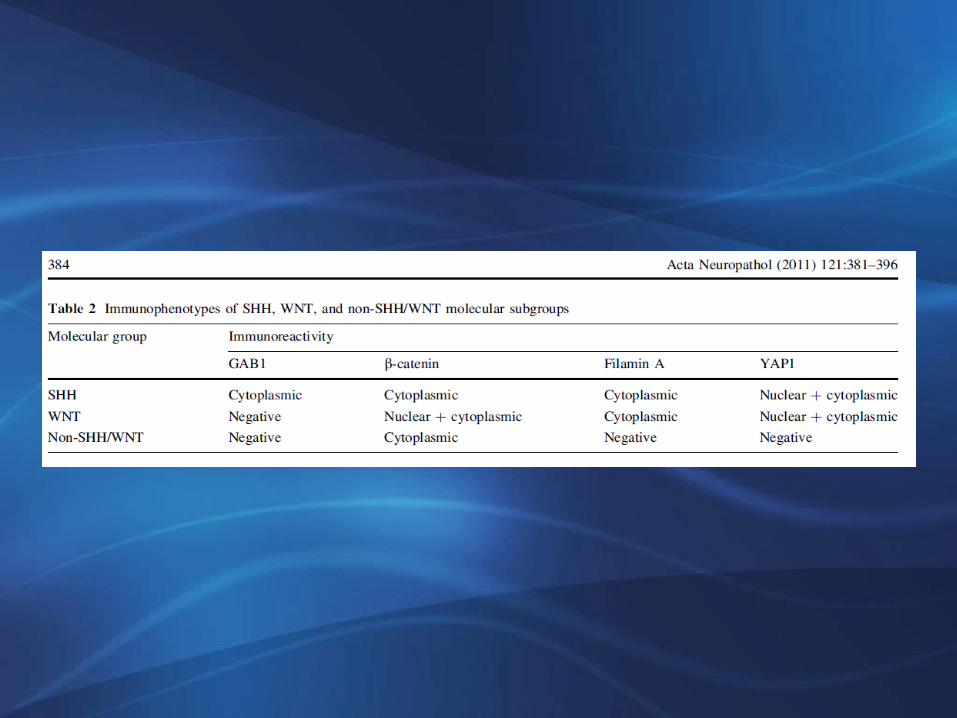
Acta Neuropathol (2006) 112: 13-22


Meduloblastomas definidos genéticamente
Meduloblastoma, WNT activadoMeduloblastoma, SHH activado y TP53-mutadoMeduloblastoma, SHH activado y TP53-nativoMeduloblastoma, no WNT/no SHH
Meduloblastoma grupo 3 (MYC)Meduloblastoma grupo 4 (i17q)

Principales Vías, PropuestaCélulas precursoras de las neuronas de la granular externa (tipo nodular / desmoplásico): Activación de la vía de Sonic hedgehog (Shh)Derivados de la zona de la matriz ventricular: Tipo clásico: Activación de la vía Winless (WNT) de meduloblastoma La mayoría de los subtipos de vía WNT de meduloblastoma son meduloblastomas clásicos, pero la mayoría de los meduloblastomas clásicos no son vía WNT ya que los tumores de vía WNT son raros




Meduloblastoma con extensa nodularidad sub grupo molecular SH
Paciente varón de 2 meses, procedente de Lima. Con TE 20 días, con vómitos y dolor.
@Sandro_CZ


Formato de reporte
1.Diagnostico integrado, incorpora toda la información
2.Clasificacion histológica3.Grado oms (refleja LA HISTORIA NATURAL)4.INFORMACION MOLECULAR


Pronóstico
En general favorable con la terapia, pero depende de la etapa y el subtipoMuy favorable, (80% de supervivencia), para la vía WNT / tipos clásicos y desmoplásicos nodulares en niños, ambos con resección totalFavorable (~ 70% de supervivencia global) para tipo Sonic hedgehog desmoplásico nodular

Pronóstico (cont)
Menos favorable para la enfermedad parcialmente resecada o metastásicaDesfavorable para tipos de células grandes / anaplásicosEspecialmente desfavorable cuando se asocia con la amplificación MYC

Pronóstico (cont)
En general, el tipo más favorable es el subtipo WNT, que es reconocido por la beta catenina nuclear y la monosomía 6La mayoría de los subtipos de vía WNT del meduloblastoma son meduloblastomas clásicos, pero no todos los meduloblastomas clásicos son la vía WNT

Comentarios
La identificación de vías moleculares (por ejemplo, WNT, Shh) se utiliza cada vez más en la clasificaciónLa separación de los grupos 3 y 4 (agrupados bajo no Shh / no WNT) no es tan importante para propósitos prácticos, actualmente difícil de hacer con inmunohistoquímica

Tumores EmbrionariosTumor embrionario con rosetas de múltiples capas C19MC alteradoTumor embrionario con rosetas de múltiples capas NOSOtros Tumores embrionarios del SNC
MeduloepiteliomaNeuroblastoma del SNCGanglioneuroblastoma del SNCTumor embrionario NOS del SNC
Tumor teratoide/rabdoide atípicoTumor embrionario del SNC con cambios rabdoides

ETANTRTumores embrionarios con rosetas verdaderas y abundante neuropil

MEDULOEPITELIOMA
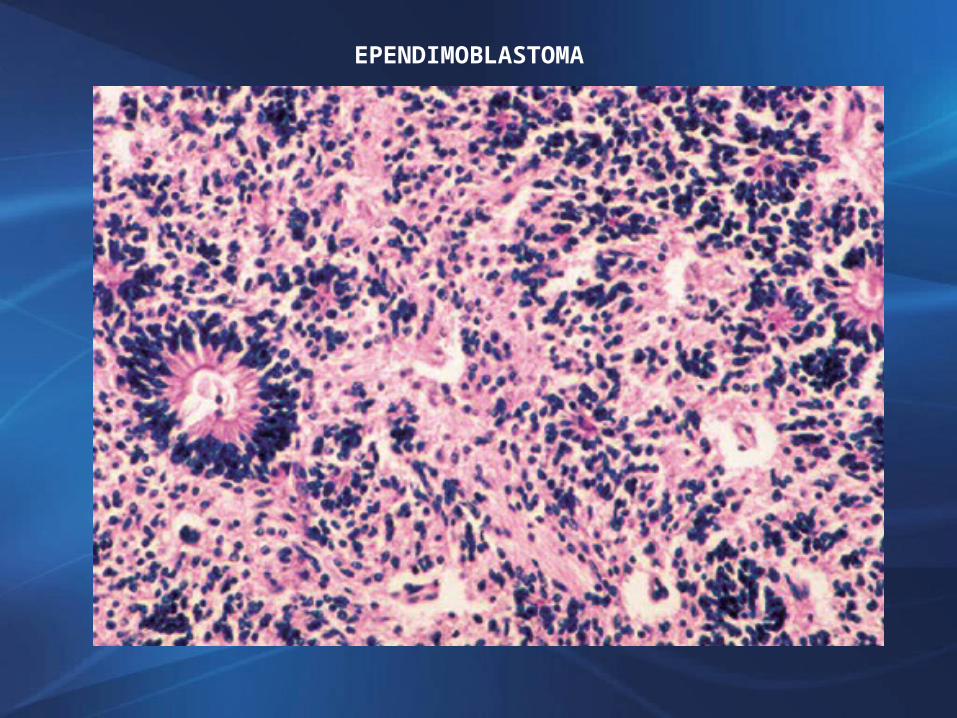
EPENDIMOBLASTOMA



Tumor embrionario con rosetas de múltiples capas C19MC alterado
Cualquier tumor embrionario del SNC con amplificación o fusión de C19MC ha sido designado ETMR, incluyendo aquellos sin rasgos histopatológicos distintivos (tales como rosetas verdaderas)

Epidemiología
Generalmente <3 añosIntracraneal, supratentorial más común que en el cerebelo, tronco encefálicoDiseminación leptomeníngea, incluso metástasis extraneurales observadas en fases posteriores de la enfermedadMal pronóstico; muy agresivo

HistologíaÁreas primitivas no estructuradas altamente celularesNeuropil finamente fibrilar paucicelularRosetas verdaderasDiferenciación focal, en áreas neuropilares, a neurocitos y células ganglionares en algunos casosRosetas delineadas por zona compuesta de uniones célula-célulaCaracterísticas de la anaplasia similar al meduloblastoma anaplásico en algunos casos

ETMRs (cont)Diagnóstico:
La sinaptofisina (+), especialmente las áreas neuropilaresAmplificación 19q13.42LIN28A (+)
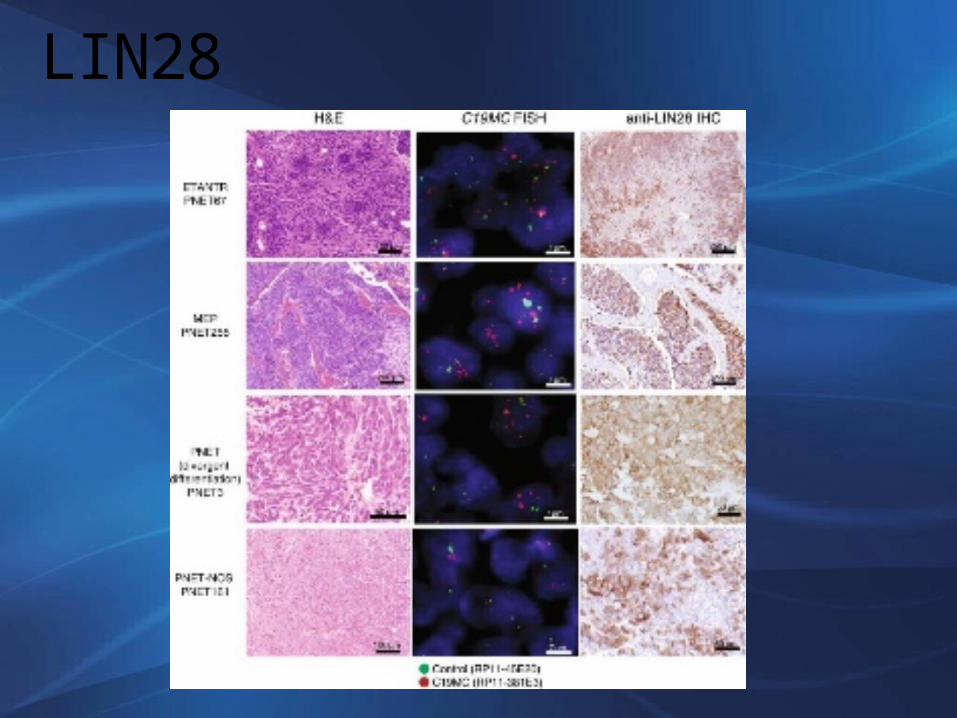
LIN28

Perlas
Abundante neuropil altamente sugestivo de la entidadLos rosetones pueden ser discretosRosetas poseen luz verdadera, no del tipo Homer Wright

Tumor teratoide rabdoide atipico
Neoplasia del SNC altamente maligna similar a tumores renales y extrarrenales que ocurren en lactantes y niños pequeños

Epidemiología
Raros: 1-2% de los tumores primarios pediátricos del SNCGeneralmente los primeros 2 años de vidaCasi todos los intracraneales; intraspinal raroPronóstico pobre

Histología
Distintivo, sin embargo, indefinido, grandes células pálidasCélulas de aspecto rabdoideCélulas indiferenciadas, simulando a veces el meduloblastomaGlándulas, rosetas o matriz condroidea (poco común)

Tumor teratoide rabdoide atipico
Varon de 4 años, procedente de Piura, con tumoración en región pineal, llega a
emergencia con vómitos explosivos, cefalea e irritabilidad

Etiología/Patogénia
Puede ocurrir esporádicamente o como parte del Síndrome de predisposición a tumores rabdoidesMutación o pérdida del locus del SMARCB1 (22q11.2)

Exámenes auxiliaresDiferenciación multilinaje, con positividad variable, a menudo en células individuales o grupos pequeñosEMA, citoqueratinas, GFAP, proteína S100, sinaptofisina, neurofilamento, cromogranina, actina del músculo liso, desminaLa pérdida de inmunoreactividad nuclear para INI1 / BAF47 esencialmente diagnóstica y requeridaLas deleciones, mutaciones en el gen INI1 / hSNF5 / SMARCB1 / BAF47 en casi todos los casos


INI-1 Inmunohistoquímica



PerlasConsiderar la posibilidad de AT / RT en cualquier tumor hipercelular, mal diferenciado en los lactantes, incluyendo embrionarios (anteriormente PNETs), meduloblastomas, carcinomas de plexo coroideoMuchos AT / RT muestran escasez de células rabdoidesAT / RT muestran pérdida difusa de INI1 inmunoticción en todas las células tumoralesLos vasos sanguíneos atrapados con INI1 nuclear positivo en las células endoteliales son control internoUn pequeño subconjunto de AT / RT no muestran pérdida de INI1 nuclear debido a la mutación en el gen SMARCA4 / BRG1La mayoría de los pacientes han heredado, mutaciones en la línea germinalPeor pronóstico que con la pérdida / mutación INI1 en el gen INI1 / hSNF5 / SMARCB1 / BAF47

Perlas (cont)No todos los tumores con pérdida / mutación INI1 en el gen INI1 / hSNF5 / SMARCB1 / BAF47 son automáticamente tumores teratoides / rabdoides atípicosOtros tumores raros del SNC tienen pérdida difusa de inmunotinción INI1: Tumor neuroepitelial cribriforme del ventrículoLos tumores raros pueden desarrollar un subconjunto de células con pérdida de INI1Astrocitomas pleomórficosGangliogliomasGliomas difusos / glioblastomasLos clones probables de células tumorales con mutaciones SMARCB1 evolucionan como evento mutacional tardíoNumerosos tumores no corrientes se reconocen ahora que tienen mutaciones INI1, es decir, tumores SMARCB1 mutadosCordomas poco diferenciados; Los típicos cordomas mantienen una fuerte inmunoreactividad nuclear INI1subconjuntos deSarcomas epitelióidesCondrosarcomas mixoides extraesqueléticosTumores epitelióides malignos del sistema nervioso periférico

Perlas (cont)No todos los tumores no AT / RT con mutaciones INI1 siguen un curso clínico altamente agresivoLos análisis transcripcionales han identificado subtipos atípicos teratoides de tumor rabdoideDiferentes patrones de expresión génicaParticipación de ASCL1, regulador de señalización NOTCHLa expresión de ASCL1, correlacionada con factores pronósticos clínicos, muestra diferencias en la supervivencia entre subgruposEjemplos raros de AT / RT sin pérdida o mutación de la proteína INI1 en el gen SMARCA4 / BRG1

NEUROBLASTOMANeoplasia de células pequeñas y anaplásicas con diferenciación neuroblástica. Se presentan en primera década de vidaHemisferios cerebralesSi presentan maduración hacia células ganglionares ganglioneuroblastomasComportamiento agresivoSingular buen pronóstico neuroblastoma olfatorio (estesioneuroblastoma) que se desarrolla sobre la placa cribiforme de la mucosa nasal
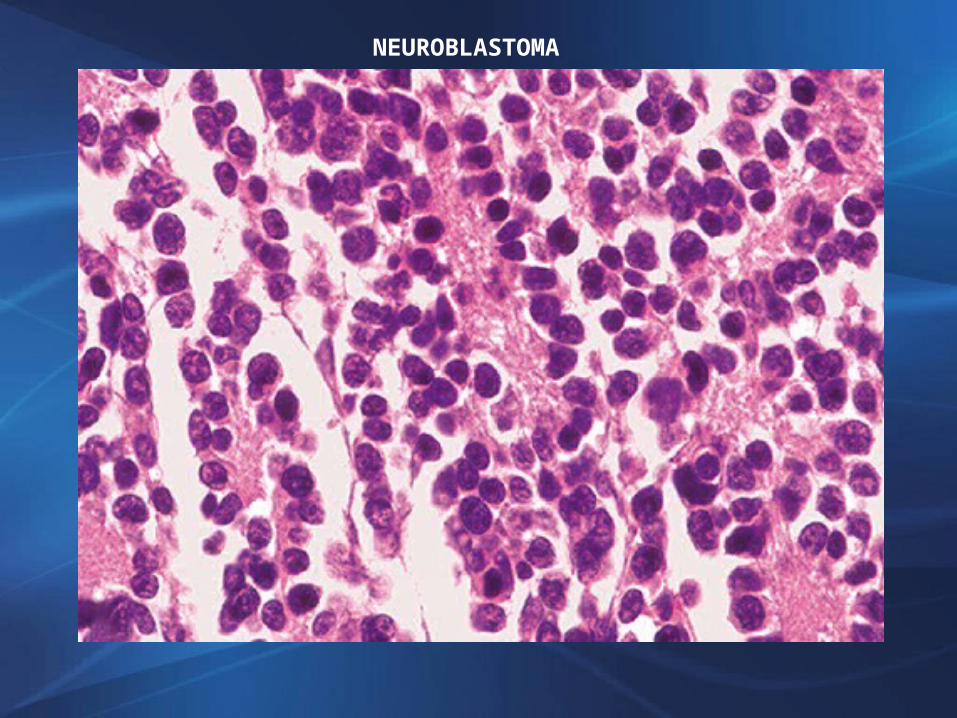
NEUROBLASTOMA

NEUROBLASTOMA

Historia Clínica:Paciente mujer nacida el 28/01/2012, quien desde los 10 meses de vida presentó vómitos en repetidas oportunidades, fiebre intermitente que no mejora con tratamiento y trastorno progresivo del nivel de conciencia. En la tomografía encefálica presenta imagen de masa heterogenea supratentorial a nivel del tercer ventrículo que condiciona hidrocefalia obstructiva, se le realiza resección completa del tumor y derivación ventrículo peritoneal, el 21/12/12.

Historia Clínica (cont):
Luego de recibir la 13° dosis de quimioterapia, se observa persistencia e incremento del tamaño de la lesión a 50x 35 mm, por lo cual se decide realizar nueva resección del tumor, el 29/04/2014.

Estudios de imagen
Tumor inicial: Imagen heterogénea predominantemente hiperintensa en T2.
Tumor post quimiterapia y radioterapia: Contenido hipointenso e hiperintenso en T1, captador de sustancia paramagnética, la tumoración se localiza en tercer ventrículo e invade ventrículo lateral.

Anatomía Patológica 1:

Anatomía Patológica 1:

Inmunohistoquímica 1:
Sinaptofisina: Positivo PGAF: Negativo

Diagnóstico 1:En el estudio anatomopatológico, se observó una neoplasia de células pequeñas, formando mantos, con tendencia Rosetoide, predominantemente perivascular, y arquitectura pseudo papilar en áreas, las células fueron de aspecto primitivo con citoplasma poco definido y escaso, la actividad mitótica es prominente. La neoplasia es positiva a Sinaptofisina. Algunas células tumorales expresaron positividad a Neurofilamento y fueron difusamente positivas a Vimentina. La PGAF y S100 fueron expresadas en escasa glía atrapada. El índice proliferativo evaluado con Ki67 es de alrededor del 40%.

TUMOR NEUROECTODERMAL PRIMITIVO DEL SNC
Conclusión Diagnóstica:

Anatomía Patológica 2:

Inmunohistoquímica 2:
Sinaptofisina: Positivo Neurofilamento: Positivo

PNET CON EXTENSA MADURACION POST TRATAMIENTO
Conclusión Diagnóstica:



Evolución:Dos meses después (20/06/2014) la paciente cursa con un cuadro convulsivo, y al ser reevaluada se identifica persistencia de la lesión a nivel de septo interventricular de 40 x 20 mm, el estudio de difusión revela escasa celularidad tumoral, ante este cuadro se decide administrar radioterapia 3600 cGY en 15 sesiones, la RMN de columna vertebral se encontró dentro de parámetros normales.La ultima RMN encefálica, post RT, mostro persistencia de la tumoración sin variación del tamaño, por lo cual se administra quimioterapia metronomica 6 ciclos, con cilcofosmamida y celecoxib.En su último control (28/08/2015). La paciente con somnolencia, deambulacion sin dificultad, y buena tolerancia oral, presenta síntomas de pubertad precoz como crecimiento mamario, y de vello púbico. RM muestra persistencia de la lesión sin variación de tamaño. Estradiol: 73.02, prolactina 6.76, TSH: 1.9, t4 1.51, FSH: 5.59 LH: 2.01, además hiponatremia probablemente secundaria SIADH.


Nueva clasificación molecular

Nueva clasificación molecular (cont)

Glioblastoma con componente neuronal primitivo
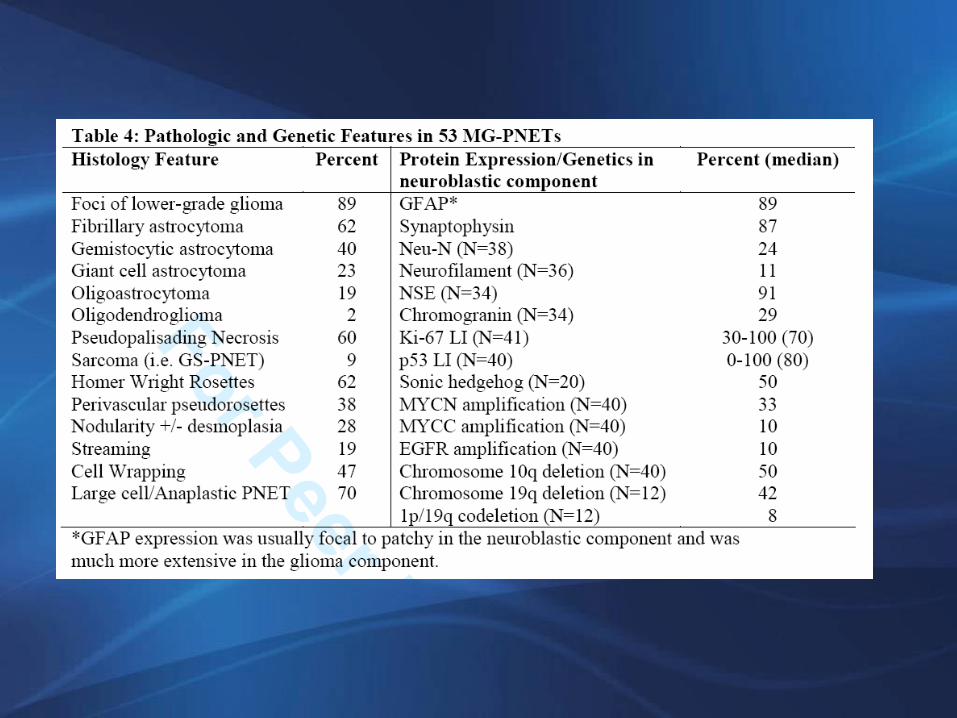

Conclusiones acerca de GBM_PNET:1. Se origina en un glioma maligno pre existente, el más
frecuente GBM2. Puede representar un proceso metaplasico o la
expansión de un clon tumoral célula madre/progenitora
3. A menudo evidencia histología anaplasica y amplificación de N-myc o C-myc
4. Tiene la capacidad de sembrar el LCR5. Puede responder a quimioterapia con regímenes de
platino

4. Comentarios finales/Plataforma de Investigación en Patología
Jefatura de BTT
1. Área de conservació
n
2. Área de laboratorios
•Lab. de Inmunohistoquimica
•Lab. Molecular•Lab. Cultivo Celular
• Lab. Corte y coloración

TUMORES DEL SNC EN BTT2006-2016
TUMORES DEL SNC EN BTT CANTIDADPORCENTAJE
(%)MENINGIOMA 115 29GLIOBLASTOMA 57 15MEDULOBLASTOMA 27 7OLIGODENDROGLIOMA 23 6SCHWANNOMA 22 6EPENDINOMA 16 4METASTASIS 12 3OLIGODENDROGLIOMA ANAPLÁSICO 12 3ASTROCITOMA DIFUSO 9 2OLIGOASTROCITOMA 6 2TUMOR DE CÉLULAS GERMINALES 6 2ASTROCITOMA PILOCÍTICO 5 1ASTROCITOMA ANAPLÁSICO 4 1OLIGOASTROCITIMA ANAPLÁSICO 4 1OTROS 75 19

29%
15%
7%6%6%
4%
3%
3%
2%2%2%1%
19%
PATOLOGÍAS NEUROLÓGICAS EN BTT2006 - 2016
MENINGIOMAGLIOBLASTOMAMEDULOBLASTOMAOLIGODENDROGLIOMASCHWANNOMAEPENDINOMAMETASTASISOLIGODENDROGLIOMA ANAPLÁSICOASTROCITOMA DIFUSOOLIGOASTROCITOMATUMOR DE CÉLULAS GERMINALESASTROCITOMA PILOCÍTICOASTROCITOMA ANAPLÁSICOOLIGOASTROCITIMA ANAPLÁSICOOTROS

Lectura recomendada

AgradecimientosResidentes de Anatomía Patológica y Equipo de Banco de tejidos Tumorales

Gracias

















![Tumores Del Snc [Autoguardado]Completo](https://static.fdocumentos.com/doc/165x107/558614e7d8b42a0e4a8b4677/tumores-del-snc-autoguardadocompleto.jpg)
