
UNIVERSIDADE FEDERAL FLUMINENSE - app.uff.br§anha, Bruna Rachel... · “Quando você pensa que...
102
UNIVERSIDADE FEDERAL FLUMINENSE FACULDADE DE FARMÁCIA PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS APLICADAS A PRODUTOS PARA SAÚDE BRUNA RACHEL DE BRITTO PEÇANHA SÍNTESE DE POLÍMEROS DE IMPRESSÃO MOLECULAR E SUA APLICAÇÃO NA TÉCNICA DE EXTRAÇÃO EM FASE SÓLIDA NITERÓI 2012
Transcript of UNIVERSIDADE FEDERAL FLUMINENSE - app.uff.br§anha, Bruna Rachel... · “Quando você pensa que...

UNIVERSIDADE FEDERAL FLUMINENSE
FACULDADE DE FARMÁCIA
PROGRAMA DE PÓS-GRADUAÇÃO EM CIÊNCIAS APLICADAS A PRODUTOS
PARA SAÚDE
BRUNA RACHEL DE BRITTO PEÇANHA
SÍNTESE DE POLÍMEROS DE IMPRESSÃO MOLECULAR
E SUA APLICAÇÃO NA TÉCNICA DE EXTRAÇÃO EM FASE SÓLIDA
NITERÓI
2012

BRUNA RACHEL DE BRITTO PEÇANHA
SÍNTESE DE POLÍMEROS DE IMPRESSÃO MOLECULAR
E SUA APLICAÇÃO NA TÉCNICA DE EXTRAÇÃO EM FASE SÓLIDA
Dissertação apresentada ao Programa de Pós-Graduação em Ciências Aplicadas a Produtos para a Saúde da Universidade Federal Fluminense, como requisito parcial para obtenção do Grau de Mestre. Área de Concentração: Pesquisa e Monitoramento de Produtos para a saúde.
Orientadoras: Profa. Dra. ESTELA MARIS FREITAS MURI
Profa. Dra. ELIANI SPINELLI
Niterói
2012

BRUNA RACHEL DE BRITTO PEÇANHA
SÍNTESE DE POLÍMEROS DE IMPRESSÃO MOLECULAR
E SUA APLICAÇÃO NA TÉCNICA DE EXTRAÇÃO EM FASE SÓLIDA
Dissertação apresentada ao Programa de Pós-Graduação em Ciências Aplicadas a Produtos para a Saúde da Universidade Federal Fluminense, como requisito parcial para obtenção do Grau de Mestre. Área de Concentração: Pesquisa e Monitoramento de Produtos para a saúde.
Aprovada em 28 de maio de 2012.
BANCA EXAMINADORA
Profa. Dra. Estela Maris Freitas Muri – OrientadoraUFF
Profa. Dra. Eliani Spinelli – OrientadoraUFF
Profa. Dra. Mônica Costa PadilhaUFRJ
Prof. Dr. Leandro Medeiros MottaUEZO
Profa. Dra. Luiza Rosaria Sousa Dias (Revisora e Suplente)UFF
Niterói2012

Dedico este trabalho à minha família,
meus pais,
meu esposo e meus filhos,
pois são a motivação que
necessito para seguir adiante.

AGRADECIMENTOS
À Deus, por me permitir mais essa conquista, tão valiosa para mim;
Às Profas. Estela e Eliani, por me aceitarem como aluna deste
projeto desafiador e pela confiança depositada em minha pessoa do
início ao fim;
Ao meu esposo Elton, sempre companheiro nos momentos difíceis,
além do suporte técnico na parte manuscrita do trabalho;
À Coordenação do PPG-CAPS, nas pessoas da Profa. Kátia e sua
secretária Adelina, pelo apoio acadêmico e disponibilidade em
atender aos alunos sempre que solicitadas;
Às Profas. Luiza Rosaria e Maria Abadia do LQMed pelo auxílio no
laboratório, sempre nos ajudando e orientando quando preciso;
À Profa. Monique Brito pela realização dos estudos de modelagem
molecular;
Às minhas amigas de mestrado e laboratório (LQMed), Bruna, Ieda,
Raquel, Simone Oliveira, Simone Pontes, Suellen e Talita pelos
momentos de descontração, carinho e solidariedade, além da troca
de preciosas idéias;
À Cássia, do Toxfree, e seu esposo Álvaro pelo apoio e ajuda com
os espectros de FTIR, de grande valia para este trabalho;
Ao prof. Venício Veiga do Instituto de Microbiologia da UFRJ por
ceder o microscópio eletrônico de transmissão para a realização das
imagens dos polímeros;
A todos que contribuíram para a realização deste trabalho.

EPÍGRAFE
“Quando você pensa que sabe todas as respostas,
vem a vida e muda todas as perguntas.”
Luís Fernando Veríssimo

RESUMO
Polímeros de impressão molecular (MIPs) foram sintetizados e aplicados como adsorventes na técnica de extração em fase sólida (EFS). O método de polimerização por precipitação foi utilizado para a síntese dos polímeros, devido à simplicidade de preparo, altos rendimentos e obtenção de partículas mais uniformes, devido a não trituração do polímero. O MIP foi sintetizado com ácido metacrílico (MAA) como monômero funcional, trimetacrilato de trimetilolpropano (TRIM) e dimetacrilato de etilenoglicol (EDMA) como agentes de reticulação e o cloridrato de amilorida (AMI) foi escolhido como molécula-molde. Diferentes proporções de MAA, TRIM, EDMA, volume e tipo de solvente foram utilizadas para ajuste das condições ideais de síntese. Os MIP foram avaliados quanto à capacidade de adsorção comparando-se a polímeros sintetizados na ausência da molécula-molde (NIP, polímeros não impressos). O solvente de elevada polaridade empregado na síntese (THF:MeOH:H2O) permitiu o emprego da técnica para moléculas polares como AMI. O controle no volume de solvente permitiu a obtenção de partículas maiores, de modo que a EFS foi realizada em condições usuais, o que confere um potencial para aplicação dessa técnica de polimerização na preparação de adsorventes para EFS. O polímero que apresentou maior capacidade adsortiva no ensaio realizado em tampão citrato-acetato pH 6,5 foi o MIP/NIP 12 (AMI:MAA:TRIM 1:8:10), com uma taxa média de adsorção de 83 e 88% para NIP e MIP, respectivamente. A adsorção foi elevada devido a interação iônica entre MAA e AMI promovida pelo controle de pH, porém foi não específica. O polímero MIP/NIP 12 foi aplicado como adsorvente na EFS, onde a recuperação de AMI foi avaliada nos resíduos de carregamento e eluição com solventes. O carregamento com tampão citrato-acetato pH 6,5 foi o ideal, favorecendo a interação iônica do polímero com o analito. A eluição total de AMI do cartucho somente ocorre após lavagem com o solvente na presença de ácido, que protona os grupos carboxila do polímero, rompendo assim a interação iônica com o analito.
Palavras-chave: Polímeros de impressão molecular (MIP). Extração em fase sólida (EFS). Polimerização por precipitação. Cloridrato de Amilorida.

ABSTRACT
Molecularly imprinted polymers (MIPs) were synthesized and applied as adsorbents in solid-phase extraction technique (SPE). The polymers have been synthesized by precipitation polymerization method because of its simplicity, high yields and good control of final size and shape of particles. MIP was synthesized using methacrylic acid (MAA) as functional monomer, trimethylolpropane trimethacrylate (TRIM) and ethyleneglycol dimethacrylate (EDMA) as cross-linker and amiloride hydrochloride (AMI) was chosen as template. Different ratios of MAA, TRIM and EDMA, volume and type of solvent were used to adjust the optimal synthesis conditions. The MIP were tested for adsorption capacity compared to the polymers synthesized in the absence of template molecule (NIP, non-imprinted polymers). The polar solvent mixture used (THF:MeOH:H2O) allowed the synthesis of MIP of polar molecules as AMI. The solvent volume control afforded the larger particles so the SPE was performed in the usual conditions, giving a potential application for this polymerization technique in the preparation of adsorbents for SPE. The polymers with higher adsorption capacity at the test performed in citrate-acetate buffer pH 6,5 was MIP/NIP 12 (AMI:MAA:TRIM 1:8:10) with adsorption rate of 83 and 88% for NIP and MIP, respectively. The recognition of MIP was due to ionic interaction between MAA and AMI promoted by pH control, but was not specific. The polymer MIP/NIP 12 was used as a solid-phase extraction sorbent and the recoveries of AMI was evaluated using different loading and elution conditions. The loading with buffer citrate-acetate pH 6,5 was optimal, due to ionic interaction of the polymer with the analyte. Total elution of AMI bound to the polymers only occurs after washing with a acid-containing solvent, because of protonation of the carboxyl groups of the polymer and disrupting the ionic interaction with the analyte.
Keywords: Molecularly imprinted polymers (MIPs); solid-phase extraction technique (SPE); precipitation polymerization method; amiloride hydrochloride.

LISTA DE SIGLAS
1-MA 1-metiladenosinaABDV Azo-bis-dimetilvaleronitrilaACN AcetonitrilaAIBN 2,2’-azo-bisisobutironitrilaALC Agente de ligação cruzada ou reticulaçãoAMI Cloridrato de amiloridaATE AtenololBPA Bisfenol ACG-EM Cromatografia gasosa acoplada à espectrometria de massasCLAE Cromatografia Liquida de Alta EficiênciaCLAE-EM Cromatografia Liquida de Alta Eficiência acoplada à espectrometria
de massas CLAE-EM/EM
Cromatografia Liquida de Alta Eficiência acoplada à espectrometria de massas em série
DBP DibutilftalatoDIP DipiridamolEDMA Dimetacrilato de etilenoglicolEFS Extração em fase sólidaFDA Food and Drug AdministrationFTIR Espectroscopia no Infravermelho com Transformada de FourierFUR FurosemidaHCT HidroclorotiazidaIR Iniciador radicalarISE Ionização por eletronsprayIUPAC Acrônimo de Internacional Union of Pure and Applied Chemistry
(Uniâo Internacional de Química Pura e Aplicada)MAA Ácido metacrílicoMeOH MetanolMET Microscopia Eletrônica de TransmissãoMF Monômero funcionalMIP Polímero de impressão molecularMISPE Extração em fase sólida com impressão molecularMM Molécula moldeNIP Polímero não impressoPB Peróxido de benzoílaPMC Perfluoro metilciclohexanoPRO PropranololPVC Policloreto de vinilaRMN Ressonância magnética nuclearSTX SaxitoxinaTHF TetrahidrofuranoTRIM Trimetacrilato de trimetilolpropanoultra-CLAE Cromatografia Líquida de Ultra-Alta EficiênciaUV UltravioletaVP 4-vinilpiridina

SUMÁRIO
1 INTRODUÇÃO, f. 15
2 OBJETIVOS, f. 19
2.1 OBJETIVO GERAL, f. 17
2.2 OBJETIVOS ESPECÍFICOS, f. 17
3 REVISÃO DA LITERATURA, f. 18
3.1 POLÍMEROS DE IMPRESSÃO MOLECULAR (MIP), f. 18
3.1.1 Síntese e variáveis experimentais, f. 18
3.1.2 Técnicas de polimerização, f. 24
3.1.3 Extração em fase sólida com impressão molecular (MISPE), f. 29
3.2 ESCOLHA DA MOLÉCULA MOLDE, f. 34
3.2.1 Estudos de modelagem molecular, f. 36
3.2.2 Propriedades físico-químicas do cloridrato de amilorida, f. 39
3.2.3 Métodos para determinação de cloridrato de amilorida, f. 41
4 MATERIAIS E MÉTODOS, f.
4.1 EQUIPAMENTOS, f. 45
4.2 REAGENTES, f. 47
4.3 PROCEDIMENTOS, f. 47
4.3.1 Síntese dos MIPs e NIPs em cloridrato de amilorida, f. 47
4.3.2 Caracterização química por Espectroscopia no Infravermelho com
Transformada de Fourier (FTIR), f. 49

4.3.3 Microscopia Eletrônica de Transmissão (MET) dos polímeros MIP/NIP 12, f.
49
4.3.4 Preparo da solução tampão citrato-acetato pH 6,5, f. 49
4.3.5 Preparo da solução estoque de AMI a 1 mg/mL, f. 50
4.3.6 Espectro de varredura de AMI em HCl 0,1N e tampão citrato-acetato pH 6,5, f.
50
4.3.7 Preparo da curva de calibração em solução tampão citrato-acetato pH 6,5, f.
50
4.3.8 Ensaio de adsorção variando a quantidade de polímero, f. 50
4.3.9 Ensaio de adsorção de AMI em MIP/NIP 7, 8, 10 e 11 variando a
concentração de AMI, f. 51
4.3.10 Ensaio de adsorção de AMI em MIP/NIP 10 e 12 variando a concentração de
AMI, f. 51
4.3.11 Ensaio de adsorção de AMI nos polímeros em THF: MeOH: água (5:4:1), f. 52
4.3.12 Confecção dos cartuchos para a EFS, f. 52
4.3.13 Procedimento da técnica de MISPE com solução de AMI, f. 52
5 RESULTADOS E DISCUSSÃO, f. 54
5.1 SÍNTESE DOS MIPs E NIPs, f. 54
5.2 CARACTERIZAÇÃO DOS POLÍMEROS OBTIDOS, f. 60
5.2.1 Caracterização química por Espectroscopia no Infravermelho com
Transformada de Fourier (FTIR), f. 60
5.2.2 Microscopia Eletrônica de Transmissão de MIP/NIP 12, f. 63
5.3 ENSAIOS DE ADSORÇÃO, f. 64
5.3.1 Ensaio de adsorção de cloridrato de amilorida em solução tampão pH 6,5, f.
64
5.3.2 Ensaio de adsorção dos polímeros em THF:MeOH:água (5:4:1), f. 72
5.4 EXTRAÇÃO EM FASE SÓLIDA COM MIP (MISPE), f. 73

6 CONCLUSÕES, f. 76
7 REFERÊNCIAS BIBLIOGRÁFICAS, f. 78
8 ANEXOS, f. 84
8.1 ESPECTROS DE FTIR DOS MONÔMEROS, f. 85
8.2 ESPECTROS DE FTIR DOS POLÍMEROS, f. 86
8.3 TABELAS COM RESULTADOS DOS ENSAIOS DE ADSORÇÃO, f. 91
8.3.1 Curva de calibração em tampão citrato-acetato pH 6,5, f. 91
8.3.2 Ensaios de adsorção com os polímeros, f. 92
8.3.3 Ensaios de adsorção variando a concentração, f. 94
8.3.4 Análise estatística com os resultados dos ensaios de adsorção (Teste t), f.
100
8.4 TABELAS COM RESULTADOS DA EXTRAÇÃO EM FASE SÓLIDA COM
MIP (MISPE), f. 101
8.5 CERTIFICADO DE ANÁLISE DO FABRICANTE DE CLORIDRATO DE
AMILORIDA, f. 102

LISTA DE FIGURAS
Figura 1. Esquema da síntese de MIP, f. 19
Figura 2. Estruturas dos principais monômeros funcionais, f. 21
Figura 3. Estruturas dos principais agentes de ligação cruzada, f. 21
Figura 4. Fases da reação de polimerização em cadeia via radicais livres, f. 22
Figura 5. Estruturas dos principais iniciadores radicalares, f. 23
Figura 6. Principais técnicas de síntese de MIP, f. 25
Figura 7. Número de publicações científicas sobre MIP usados como sistemas de liberação de fármacos nos últimos anos, f. 29
Figura 8. Técnica de Extração em Fase Sólida, f. 30
Figura 9. Compostos candidatos a molécula molde para a STX, f. 37
Figura 10. Modificações na estrutura da amilorida sugeridas na modelagem molecular, f. 38
Figura 11. Estrutura da amilorida, f. 40
Figura 12. Esquema da síntese de MIP de AMI com EDMA e MAA, f. 60
Figura 13. Espectros de FTIR de MIP e NIP 7 (EDMA), f. 61
Figura 14. Espectros de FTIR de MIP e NIP 11 (TRIM), f. 62
Figura 15. Espectro de FTIR de AMI, f. 63
Figura 16. Imagens de MET dos polímeros NIP e MIP, f. 64
Figura 17. Espectro de varredura de cloridrato de amilorida, f. 65
Figura 18. Curva de calibração AMI em tampão citrato-acetato pH 6,5, f. 65
Figura 19. Gráficos de adsorção de acordo com a massa de polímero, f. 66
Figura 20. Curvas de adsorção de MIPs/NIPs 7, 8, 10, 11 e 12, f. 68
Figura 21. Curvas de adsorção de MIP/NIP 10 e 12 de acordo com a concentração, f. 69
Figura 22. Interação iônica de amilorida com o ácido metacrílico no pH 6,5, f. 71

LISTA DE TABELAS
Tabela 1. Compostos selecionados para os estudos de modelagem, f. 36
Tabela 2. Quantidades dos reagentes na síntese dos MIPs e NIPs de cloridrato de amilorida, f. 48
Tabela 3. Rendimentos das reações de síntese dos MIPs e NIPs, f. 55
Tabela 4. Aspecto dos polímeros obtidos, f. 56
Tabela 5. Taxas médias de adsorção (%) dos polímeros, f. 70
Tabela 6. Recuperação de AMI (%) obtida pela técnica de MISPE, f. 74

1 INTRODUÇÃO
Os polímeros de impressão molecular (Molecularly Imprinted Polymers - MIP)
consistem em matrizes macromoleculares sintéticas, obtidas com base em métodos
de moldagem molecular e que são capazes de reconhecer seletivamente moléculas
biológicas, como fármacos e proteínas. A especificidade na detecção de substâncias
é comparável à dos anticorpos monoclonais utilizados em técnicas imunológicas
(SOUSA; BARBOSA, 2009).
Para sintetizar o MIP, uma molécula molde é colocada para interagir com as
moléculas de um monômero. A molécula molde pode ser o próprio analito ou um
composto com estrutura semelhante ao mesmo. O monômero, denominado
funcional, deve possuir grupos que possibilitem a interação com a molécula molde,
sendo que esta pode ser de natureza covalente ou não covalente. Adiciona-se ao
meio, um segundo monômero (estrutural) capaz de promover ligações cruzadas
entre os monômeros primários, a fim de formar uma matriz polimérica rígida. A
reação de polimerização é do tipo em cadeia e começa após a adição de um
iniciador radicalar, espécie química capaz de liberar radicais livres para iniciar a
reação. Por fim, a molécula molde é removida da matriz polimérica por meio de
solvente ou clivagem química (no caso de ligação covalente). Assim, espera-se que
o polímero resultante contenha microcavidades com tamanhos uniformes capazes
de reter seletivamente a molécula molde em uma amostra complexa, eliminando
também possíveis interferentes (TARLEY; SOTOMAYOR; KUBOTA, 2005a).
Além do caráter seletivo, os MIPs demonstram vantagens em relação aos
adsorventes de natureza biológica quanto à estabilidade química, capacidade de
adsorção e reprodutibilidade no preparo do polímero, além de menor custo,

16
preparação rápida, resistência térmica e melhores condições de estocagem
(TARLEY; SOTOMAYOR; KUBOTA, 2005a e b).
O emprego de MIP como material adsorvente na técnica de extração em fase
sólida (EFS) vem adquirindo destaque, pois oferece alto grau de seletividade quando
comparados com outros materiais, como a sílica modificada (C18) e resinas de troca
iônica e, ao mesmo tempo, por serem mais estáveis que os adsorventes de origem
biológica (imunosorventes). A EFS consiste na percolação da amostra no material
adsorvente disposto em cartuchos, onde a espécie de interesse fica retida. Quando
o material adsorvente é um MIP, a técnica recebe a designação de Molecular
Imprinting Solid-Phase Extraction (MISPE) (TARLEY; SOTOMAYOR; KUBOTA,
2005a).
A técnica de EFS promove a eliminação dos principais interferentes presentes
na matriz, principalmente de natureza biológica, possibilitando assim a obtenção de
um resíduo mais limpo. Com o uso de um pequeno volume de solvente para eluir o
analito do cartucho promove-se a sua concentração. Além da economia com
reagentes e solventes, o tempo gasto com extração e purificação é menor. A EFS é
usualmente empregada para o preparo de amostras de urina visando a análise
toxicológica (COSTA, 2008).
A técnica de extração em fase sólida representa uma estratégia interessante
para a extração de substâncias, em particular as de elevada polaridade, em matrizes
de natureza altamente hidrofílica como as biológicas. Na análise de substâncias
polares em urina, por exemplo, a extração líquido-líquido é a técnica mais utilizada,
e geralmente fornece rendimentos baixos devido a solubilidade destas na fase
aquosa. Além das propriedades da EFS, a MISPE acrescenta um aumento na
seletividade na extração do analito de interesse frente a outros compostos presentes
na amostra, devido ao efeito de impressão molecular, além de reduzir os efeitos dos
interferentes da matriz da amostra (BLOMGREN, et al., 2002).

2 OBJETIVOS
2.1 OBJETIVO GERAL
O objetivo geral do projeto consiste na síntese de Polímeros de Impressão
Molecular (MIPs) para serem utilizados como adsorventes na técnica de extração
em fase sólida.
2.2 OBJETIVOS ESPECÍFICOS
2.2.1 Escolha da molécula molde a ser usada na síntese dos Polímeros de
Impressão Molecular (MIPs);
2.2.2 Síntese dos MIPs com a molécula molde escolhida no item 2.2.1 e seus
polímeros não impressos (NIP) correspondentes;
2.2.3 Caracterização dos polímeros obtidos (MIPs e NIPs);
2.2.4 Ensaio de adsorção do analito escolhido nos MIPs e NIPs sintetizados e
avaliação da capacidade adsortiva dos mesmos;
2.2.5 Confecção do cartucho para a EFS com os polímeros de maior capacidade
adsortiva (MISPE);
2.2.6 Procedimento da técnica de MISPE com solução padrão para determinação
da recuperação do analito em MIP e NIP.

3 REVISÃO DA LITERATURA
3.1 POLÍMEROS DE IMPRESSÃO MOLECULAR (MIP)
3.1.1 Síntese e variáveis experimentais
A impressão molecular é uma tecnologia capaz de produzir polímeros dotados
de sítios específicos de reconhecimento, moldados a partir de uma molécula molde
(MM), sendo esta o próprio analito ou um composto de estrutura semelhante.
Nesses polímeros, rotineiramente denominados de Polímeros de Impressão
Molecular (Molecularly Imprinted Polymers, MIP), a etapa de síntese ocorre após a
formação de um complexo entre o monômero funcional (MF) e a MM. Assim, as
terminações ligantes dos MF são posicionadas em pontos complementares àqueles
provenientes da MM, permitindo a formação de ligações. Após a adição de um
agente de ligação cruzada (“cross-linker”) e um reagente iniciador, a reação de
polimerização tem início e o MIP é formado (Figura 1), ocorrendo a formação de
uma matriz polimérica rígida (FIGUEIREDO; DIAS; ARRUDA, 2008; CORMACK;
ELORZA, 2004).
Em seguida, após a extração da molécula molde (também referida como
molécula chave, molécula alvo, molécula impressa, analito ou template, do inglês),
realizada por meio de um solvente adequado, são formadas cavidades
complementares a este molde em tamanho, forma e presença de grupos funcionais
(TARLEY; SOTOMAYOR; KUBOTA, 2005a; TAMAYO; TURIEL; MARTÍN-
ESTEBAN, 2005). Tais cavidades constituem os sítios específicos de ligação que
agem no reconhecimento deste composto e/ou de substâncias com estrutura
semelhante a ele. Por causa desta capacidade, os MIPs podem ser conhecidos
como anticorpos plásticos, já que os mesmos podem atuar como biomiméticos,

19
mimetizando o mecanismo de ação de enzimas ou a interação fármaco-receptor
(YAN; RAMSTRÖM, 2005).
Figura 1. Esquema da síntese de MIP
Fonte: TARLEY; SOTOMAYOR; KUBOTA, 2005a
O primeiro MIP foi sintetizado em 1972, quando Wulf e Sarhan descreveram a
síntese de um polímero com sítios seletivos para a separação enantiomérica de
racematos de açúcares (WULF; SARHAN, 1972). Desde então, estes materiais têm
recebido grande atenção por causa das vantagens, em relação aos materiais
biológicos, que atuam no mecanismo de reconhecimento molecular. A facilidade de
síntese, o baixo custo dos reagentes empregados, a estabilidade química, física e
térmica do material por longos períodos de tempo, a capacidade de ser estocado
sem perder sua especificidade e a possibilidade de reutilização do polímero, após a
limpeza do mesmo, são algumas dessas vantagens (MAIER et al., 2004; MAHONY
et al., 2005).
Em contrapartida, os materiais imunosorventes possuem algumas
desvantagens como o elevado custo, a variação de sua estabilidade, a fragilidade
química, física e térmica dos mesmos, a necessidade de condições especiais de
armazenagem, a dificuldade de obtenção de biomoléculas e instabilidade quando

20
estes são empregados em condições diferentes daquelas presentes em seu
ambiente nativo (MAIER et al., 2004; TARLEY; SOTOMAYOR; KUBOTA, 2005b).
Desta forma, os MIPs têm sido empregados como substitutos de biomoléculas que
atuam no processo de reconhecimento molecular (MAIER et al., 2004; BAGGIANI et
al., 2001). Entretanto, os Polímeros de Impressão Molecular apresentam algumas
desvantagens como a baixa capacidade catalítica e a natureza polidispersa dos
sítios de reconhecimento, embora isto não seja sempre uma limitação na prática.
Dependendo de como o MIP é sintetizado, a quebra e trituração do material torna o
processo laborioso e pode haver a dificuldade de remoção da molécula molde em
alguns casos (MAHONY et al., 2005).
Em todos os processos de moldagem molecular, a MM assume uma
importância fundamental, já que é responsável pela definição da organização
espacial dos grupos funcionais dos MF. Os MF são responsáveis pelas interações
que se estabelecem nos locais de reconhecimento. É muito importante assegurar a
complementaridade da funcionalidade da MM com a do MF (por exemplo, um
doador de prótons com um aceptor de prótons), de modo a maximizar a formação
dos complexos e, assim, o processo de moldagem molecular do polímero (SOUZA;
BARBOSA, 2009).
Entre os MF mais empregados para o preparo dos MIPs (Figura 2), está o
ácido metacrílico (MAA, 1). No entanto, deve-se salientar que a escolha deste é feita
de acordo com a natureza do analito. A maioria deles possuem a capacidade de
fazer ligações de hidrogênio e interação iônica com o analito. Os analitos que
possuem grupos básicos, por exemplo, interagem mais facilmente com monômeros
que contenham grupos ácidos, como o MAA e monômeros com caráter básico,
como o 4-vinilpiridina (4-VP, 2), interagem preferencialmente com analitos ácidos. A
acrilamida (3) faz interações com o analito somente através de ligações de
hidrogênio (TARLEY; SOTOMAYOR; KUBOTA, 2005a).
O agente de ligação cruzada ou reticulação (ALC) usado na obtenção de um
MIP desempenha três funções principais: controle da morfologia da matriz polimérica
(polímero do tipo gel, polímero macroporoso ou pó microgel), estabilização dos
locais de ligação com capacidade de reconhecimento molecular e estabilização
mecânica da matriz polimérica. Da mesma forma que ocorre a interação entre a MM
e o MF, o ALC deve ter grupos que possam interagir de maneira estável com o MF
para a formação da matriz polimérica.
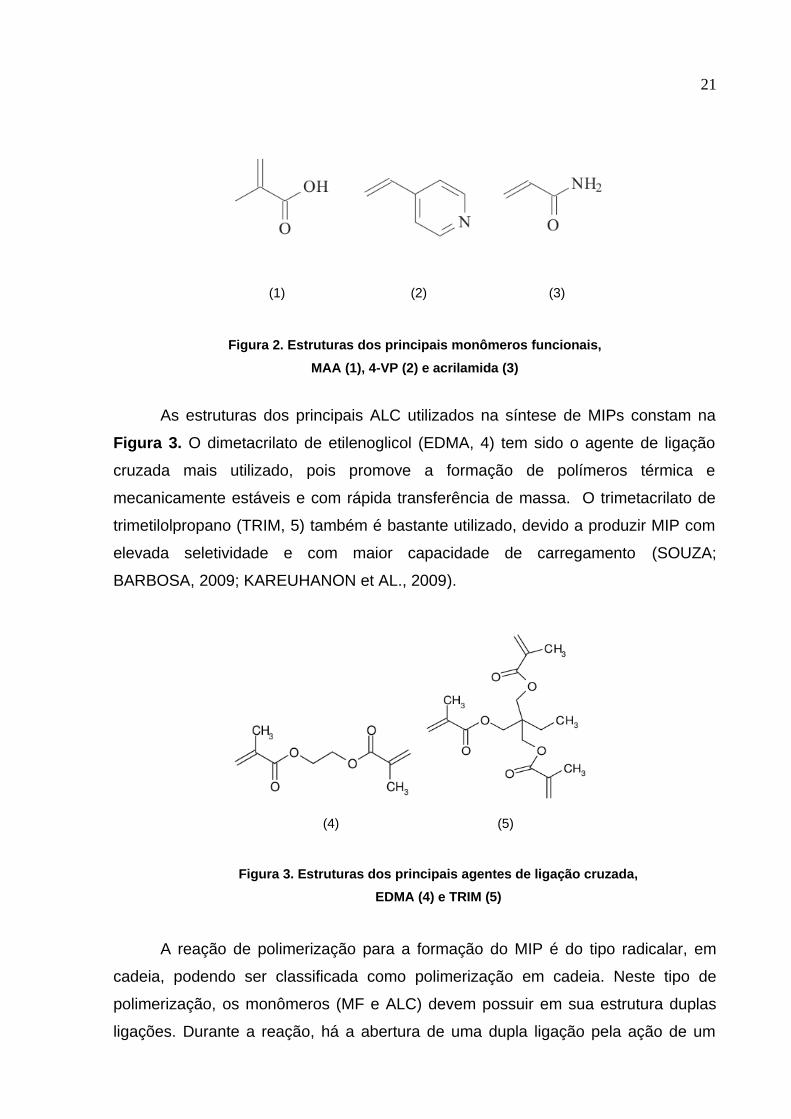
21
(1) (2) (3)
Figura 2. Estruturas dos principais monômeros funcionais,
MAA (1), 4-VP (2) e acrilamida (3)
As estruturas dos principais ALC utilizados na síntese de MIPs constam na
Figura 3. O dimetacrilato de etilenoglicol (EDMA, 4) tem sido o agente de ligação
cruzada mais utilizado, pois promove a formação de polímeros térmica e
mecanicamente estáveis e com rápida transferência de massa. O trimetacrilato de
trimetilolpropano (TRIM, 5) também é bastante utilizado, devido a produzir MIP com
elevada seletividade e com maior capacidade de carregamento (SOUZA;
BARBOSA, 2009; KAREUHANON et AL., 2009).
(4) (5)
Figura 3. Estruturas dos principais agentes de ligação cruzada,
EDMA (4) e TRIM (5)
A reação de polimerização para a formação do MIP é do tipo radicalar, em
cadeia, podendo ser classificada como polimerização em cadeia. Neste tipo de
polimerização, os monômeros (MF e ALC) devem possuir em sua estrutura duplas
ligações. Durante a reação, há a abertura de uma dupla ligação pela ação de um

22
iniciador que, no caso da síntese de MIP, é um composto capaz de produzir radicais
livres, denominado iniciador radicalar. As fases da reação de polimerização por
adição são a iniciação, a propagação e o término (Figura 4). O início da formação
da cadeia polimérica então se dá início a partir do rompimento da dupla ligação de
um monômero pela ação de um radical livre formado por cisão homolítica do
iniciador, produzindo um novo radical (CANEVAROLO Jr., 2006).
Iniciação
R1OOR1 → 2 R1OO·
R2 – HC=CH – R3→ R2– HC – CH – R3·
2 R1OO· + R2– HC – CH – R3 · → R1OO – R3– HC – CH – R2·Propagação
R1OO – R3– HC – CH – R2· + R2– HC – CH – R3 · → R4·Término
R4· + R5· → R4 – R5 (combinação)
R4· + R5· → R4 + R5 (desproporcionamento)
R4· + R5 → R4 + R5· (transferência de cadeia)
R4· + S → R4 + S· (transferência para o solvente)
Figura 4. Fases da reação de polimerização em cadeia via radicais livres
A fase de propagação é a reação deste radical com outra ligação dupla de
outra molécula de monômero, e assim sucessivamente, produzindo macrorradicais.
O término da formação das cadeias poliméricas pode ocorrer através da combinação
de dois macrorradicais em crescimento, por desproporcionamento, onde dois
macrorradicais se rearranjam e param o crescimento das duas cadeias, por
transferência de cadeia (uma cadeia em crescimento transfere o radical para uma
cadeia terminada, fazendo esta crescer novamente e terminando a primeira) ou
transferência para o solvente, onde o radical da cadeia em crescimento é transferido
para o solvente (CANEVAROLO Jr., 2006).

23
A função do iniciador radicalar (IR) é liberar radicais livres para possibilitar o
início e a manutenção da reação de polimerização. Contudo, para o início da reação,
é necessário algum estímulo físico como o aumento da temperatura ou a incidência
de radiação UV. Esse estímulo é determinante na escolha do IR, pois os outros
reagentes da síntese (MM, MF, ALC) podem ser termo ou fotossensíveis. As
estruturas dos principais IRs utilizados em impressão molecular estão na Figura 5.
O IR 2,2’-azo-bisisobutironitrila (AIBN, 6) é o mais empregado na síntese dos MIPs,
mas outros também podem ser utilizados, como azo-bis-dimetilvaleronitrila (ABDV,
7) e peróxido de benzoíla (PB, 8) (FIGUEIREDO; DIAS; ARRUDA, 2008;
CORMACK; ELORZA, 2004; KAREUHANON et al., 2009).
(6) (7) (8)
Figura 5. Estruturas dos principais iniciadores radicalares,
AIBN (6), ABDV (7) e PB (8)
O solvente na síntese do MIP deve ser cuidadosamente escolhido de acordo
com a natureza da molécula molde e dos reagentes. Além de dissolver os reagentes
da síntese, este não deve interferir na formação do complexo MM-MF, o que pode
resultar na formação de sítios de ligação pouco seletivos e em pequeno número. A
maioria dos MIPs é sintetizada em solventes de polaridade mais baixa, apróticos e
de baixa constante dielétrica, como por exemplo, clorofórmio e tolueno (MATSUI et
al., 1995; HAGINAKA et al., 1999). Entretanto, alguns MIPs já foram sintetizados
com sucesso em solventes polares como metanol (CAI; GUPTA, 2004) e acetonitrila
(THEODORIDIS; MANESIOTIS, 2002), além de misturas de solventes como THF,
metanol e água visando a aplicação do MIP em análises de amostras aquosas,
como alimentos e fluidos biológicos (KAREUHANON et al., 2009).
Existem vários processos de moldagem molecular, permitindo gerar locais
com elevada capacidade e especificidade de reconhecimento molecular, que se

24
distinguem pela natureza da ligação estabelecida com a MM, quer durante o
processo de síntese do polímero, quer na fase de recaptação da MM. A moldagem
covalente se caracteriza pelas ligações covalentes que se formam entre MM e MF,
com estequiometria definida, mas com cinética menos favorável na recaptação do
analito. Essas ligações podem ser reversíveis ou irreversíveis, o que implica para
este último em condições mais drásticas para remoção do template sem
recuperação deste em sua forma original, além da recaptação do mesmo ser
estericamente desfavorável. Na moldagem não covalente, as interações podem ser
através de ligações de hidrogênio ou de natureza iônica, além de interações
hidrofóbicas e dipolo-dipolo (WHITCOMBE; VULFSON, 2001). Neste processo, a
MM pode ser facilmente extraída do polímero e reciclada, o que é particularmente
interessante pela sua simplicidade, permitindo a obtenção de MIP que apresentam
uma afinidade elevada para o seu alvo e uma ampla aplicabilidade. (SOUSA;
BARBOSA, 2009).
A técnica de impressão molecular envolve a preparação em paralelo ao MIP
de um polímero não impresso (NIP), ou polímero controle, ou seja, o MIP sintetizado
sem a presença da molécula molde. A formação das cavidades complementares em
MIP pela interação com o analito pode ser verificada pela maior retenção deste no
polímero impresso, comparado ao NIP, em determinadas condições de análise que
devem ser otimizadas para cada composto. Em NIP, ocorre a formação de sítios não
seletivos, de forma aleatória, desprovidos de cavidades complementares ao volume
e posição de grupos que podem interagir com o analito, promovendo alguma
retenção do mesmo.
3.1.2 Técnicas de polimerização
Diferentes usos e potenciais aplicações dos MIPs exigem diferentes
propriedades dos polímeros. Alguns fatores tais como especificidade, capacidade ou
o meio em que se encontra a amostra e o analito requerem características
particulares nos MIPs. Em resposta a essa demanda, diferentes métodos para
produzir polímeros impressos foram desenvolvidos. As técnicas mais empregadas
para o preparo de MIP são a polimerização em massa, por suspensão, por
precipitação, polimerização por expansão em multietapas (“multi-step swelling”) e
por emulsão (Figura 6). Cada um desses procedimentos envolve o controle de

25
diferentes parâmetros durante a síntese e produz polímeros com propriedades
diferentes (PÉREZ-MORAL; MAYES, 2004).
Figura 6. Principais técnicas de síntese de MIP: (A) Massa (B) Suspensão
(C) Precipitação (D) Expansão em multietapas e (E) Emulsão (“Core-shell emulsion”)
Fonte: Adaptado de PÉREZ-MORAL; MAYES, 2004
Os MIPs são preparados convencionalmente pelo método de polimerização
em massa (ou bulk, do inglês), onde a reação é realizada em sistema homogêneo.
Esta reação é conduzida em frascos selados contendo monômero, molécula molde,
solvente, agente de ligação cruzada e iniciador radicalar. A reação ocorre na
ausência de oxigênio sob fluxo de N2 ou argônio e induzida com aquecimento e/ou
radiação UV. O oxigênio deve ser eliminado do meio reacional, pois retarda a reação
de polimerização radicalar. Por fim, o sólido polimérico resultante é moído,
peneirado e submetido a uma lavagem com solvente para extração do analito,
visando seu uso posterior. As partículas obtidas por este processo possuem forma
irregular, com o tamanho variando entre 20 e 50 µm. Uma outra desvantagem é que
uma grande parte do polímero produzido (cerca de 50%) perde suas propriedades
analíticas, uma vez que durante a trituração, alguns sítios seletivos formados são
parcialmente destruídos, diminuindo a capacidade seletiva de retenção do polímero.
Também pode ocorrer a formação de áreas de heterogeneidade na matriz
polimérica, resultante da falta de controle do processo durante a polimerização,

26
particularmente quando a iniciação da polimerização é feita por UV. Os MIPs obtidos
pelo método de polimerização em massa são mais indicados para técnicas de
extração como a EFS (partículas da ordem de micrômeros), ao passo que seu uso
como fase estacionária em CLAE é pouco indicado, devido à heterogeneidade das
partículas (TARLEY; SOTOMAYOR; KUBOTA, 2005a; PÉREZ-MORAL; MAYES,
2004).
A polimerização por suspensão tem como objetivo a obtenção de
microesferas com maior homogeneidade de tamanho. A reação polimérica ocorre
dentro das gotas do monômero dispersas num solvente imiscível, geralmente água.
Os reagentes da síntese (MF, MM, ALC e IR) são dissolvidos em solvente orgânico
e, em seguida, adicionados na fase dispersiva (polar), sendo a fase apolar voltada
para o interior da micela, normalmente estabilizada pelo emprego de um tensoativo,
geralmente o álcool polivinílico ou polímeros à base de sais orgânicos
(FIGUEIREDO; DIAS; ARRUDA, 2008). A reação de polimerização ocorre após o
aquecimento do meio reacional sob agitação. O polímero obtido no formato de
esferas é facilmente separado da fase aquosa. Apesar da uniformidade do tamanho
das partículas ser uma vantagem, algumas moléculas de água se dissolvem na
mistura polimérica interferindo na interação analito-monômero, principalmente
quando esta se dá por ligação de hidrogênio. Além disso, quando monômeros e
analitos possuem elevada solubilidade em água, pode ocorrer partição destes
constituintes da fase orgânica para o meio aquoso, resultando num polímero com
baixo reconhecimento molecular. A polimerização por suspensão é mais adequada
para analitos e monômeros que interagem preferencialmente por interações
hidrofóbicas e iônicas (TARLEY; SOTOMAYOR; KUBOTA, 2005a).
A fim de solucionar a limitação constatada no método de polimerização por
suspensão em meio aquoso, foi desenvolvida uma reação de polimerização em
suspensão empregando o agente dispersante perfluorocarbono (perfluoro
metilciclohexano - PMC). Este agente é imiscível em muitos compostos orgânicos e,
consequentemente, permite a formação de uma fase inerte apropriada para a
polimerização. Além das vantagens em relação ao método de polimerização por
suspensão em meio aquoso no que tange à influência da água, no método com o
emprego de PMC é possível também controlar o tamanho das partículas poliméricas
(de 5 a 50 μm) alterando a massa do dispersante (TARLEY; SOTOMAYOR;
KUBOTA, 2005a).

27
O método de preparo de MIP por meio de polimerização por precipitação é
análogo ao método de polimerização em massa, porém, volumes superiores de
solventes são empregados. Enquanto que, no método convencional normalmente
são utilizados volumes de 10 mL, no método por precipitação são usados cerca de 5
vezes mais. Neste método não é necessário usar nenhum tipo de estabilizante para
evitar a coagulação das gotas de monômero. A formação de partículas poliméricas
ocorre devido ao crescimento da cadeia polimérica ao longo da reação, o que torna
o polímero cada vez menos solúvel no meio. Além disso, a precipitação também
ocorre em face da formação de ligações cruzadas no polímero, tornando-o também
pouco solúvel no solvente reacional. Devido a estes fatores, a formação de
microesferas na forma de um precipitado é assegurada sem a necessidade de se
empregar agentes estabilizantes. A polimerização por precipitação também produz
partículas com tamanhos pequenos e uniformes (diâmetro entre 0,3 – 10 μm) com
maior rendimento da reação (85%). A síntese dos MIPs por polimerização por
precipitação é induzida por luz ou aquecimento na ausência de oxigênio, quando
ambos, monômero e analito são dissolvidos no solvente, seguido da adição do
reagente de ligação cruzada e iniciador radicalar. Neste método as micropartículas
são obtidas por meio de centrifugação. O método de polimerização por precipitação
produz partículas com diâmetros reduzidos e essa característica torna-se uma
desvantagem quando tais polímeros são utilizados como fases estacionárias em
CLAE, dada a elevada pressão resultante dentro da coluna cromatográfica
(TARLEY; SOTOMAYOR; KUBOTA, 2005a).
O método de polimerização por expansão em multietapas é o mais trabalhoso
e envolve basicamente o emprego de partículas poliméricas com diâmetro bem
definido (geralmente poliestireno com diâmetro de 1 μm). Diferente de outros
métodos, as partículas poliméricas atuam como solventes porogênicos, permitindo a
produção de polímeros com tamanhos e poros maiores. Primeiramente, uma
microemulsão de partículas de poliestireno é preparada em água contendo o
tensoativo dodecil sulfato de sódio e o solvente dibutilftalato. O sistema é agitado até
eliminar a microemulsão. Em seguida, a expansão das partículas de poliestireno é
efetuada após adição de uma microemulsão preparada a partir do iniciador radicalar,
solvente porogênico e álcool polivinílico como agente estabilizante. Após um
determinado tempo de agitação, o analito junto com o monômero funcional e o
reagente de ligação cruzada dispersos em água, além de álcool polivinílico, são

28
adicionados ao meio. O sistema é novamente agitado, caracterizando a segunda
etapa de expansão das partículas de poliestireno. Por fim, a reação de
polimerização do MIP é iniciada sob agitação e induzida por aquecimento sob
atmosfera inerte. As partículas monodispersas do MIP são posteriormente
separadas do sobrenadante e submetidas a uma lavagem com solvente para
extração do analito, e se apresentam com diâmetro entre 2 – 50 μm (TARLEY;
SOTOMAYOR; KUBOTA, 2005a).
O método de polimerização por emulsão envolve a formação de um núcleo
polimérico, primeiramente, em fase aquosa contendo os monômeros e iniciador,
adicionada de tensoativos e com controle de temperatura e agitação. As partículas
obtidas são filtradas e colocadas para reagir em nova fase aquosa com tensoativo,
monômeros, iniciador e o analito. As partículas de MIP vão se formando ao redor do
núcleo, em forma de concha (daí a denominação “core-shell”). As partículas obtidas
por este método são monodispersas e podem ser produzidas em um intervalo de
tamanho coloidal de 0,05 – 2 μm (PÉREZ-MORAL; MAYES, 2004).
A aplicação da tecnologia de impressão molecular é muito ampla, onde
existem estudos na área da química analítica em métodos de extração (extração e
microextração em fase sólida), separação (como adsorventes em cromatografia
líquida) e no desenvolvimento de sensores químicos, na área da química sintética
(como catalisadores), na indústria de alimentos (para remoção de cafeína e
colesterol de amostras) e na biotecnologia, como material de suporte para
crescimento de células e para purificação de proteínas (WHITCOMBE; VULFSON,
2001). Mais recentemente, os MIP têm sido estudados para serem utilizados como
uma ferramenta para o desenvolvimento de sistemas de liberação de fármacos
(“DDS – drug delivery systems”), aumentando o número de publicações nas últimos
anos, como pode ser visto na Figura 7.
A aplicabilidade dos MIPs em diferentes segmentos da química analítica faz
com que a adoção dos procedimentos de preparo seja dependente das
características intrínsecas das técnicas. Significa, portanto, que o preparo de um
MIP visando seu uso em técnicas de separação é diferente daquele onde o MIP vai
ser utilizado na modificação de eletrodos ou sensores químicos (TARLEY;
SOTOMAYOR; KUBOTA, 2005b).

29
Figura 7. Número de publicações científicas sobre MIPs
usados como sistemas de liberação de fármacos nos últimos anos.
Fonte: FIGUEIREDO; DIAS; ARRUDA, 2008
3.1.3 Extração em fase sólida com impressão molecular (MISPE)
O emprego dos MIPs como materiais adsorventes em EFS vem adquirindo
destaque, pois oferecem alto grau de seletividade quando comparado com outros
adsorventes, como a sílica modificada (C18) e resinas de troca iônica e, ao mesmo
tempo, por serem mais estáveis que os imunosorventes. Um procedimento de EFS
consiste na percolação da amostra no material sorvente disposto em cartuchos,
onde a espécie de interesse fica retida seletivamente quando do uso do MIP. Em
seguida, ao se empregar amostras complexas, faz-se necessário efetuar uma etapa
de limpeza com um solvente adequado, no intuito de extrair as espécies
interferentes ligadas ao polímero por interações não específicas. Nesta etapa, a
espécie de interesse não deve ser coeluída. As interações não específicas formadas
durante a síntese, responsáveis pela retenção das espécies interferentes no MIP são
originadas devido ao excesso de monômeros no meio reacional. Sendo assim, na
extração em fase sólida normalmente é realizada uma etapa de limpeza também
conhecida como etapa de lavagem, a fim de extrair os interferentes retidos nos sítios
não específicos. A etapa final é efetuada mediante a eluição do analito na ausência
dos interferentes. Nestes procedimentos, as etapas de limpeza e de pré-
concentração são executadas simultaneamente, o que confere também aos MIPs

30
características de materiais pré-concentradores, como visto na Figura 8 (TARLEY;
SOTOMAYOR; KUBOTA, 2005a; PEREIRA, 2008).
Figura 8. Técnica de extração em fase sólida. Em (1), a amostra é adicionada ao cartucho
contendo o MIP e o analito se liga seletivamente ao polímero (2). Em (3) ocorre a eluição dos
componentes da matriz e posterior eluição do analito com solvente apropriado.
Fonte: ANDERSSON, L.I., 2000
A análise de microcistina-LR através de MISPE foi descrita por Chianella et al.
(2003). O uso da técnica permitiu a concentração da amostra em até 1000 vezes,
facilitando a quantificação em água potável, onde os níveis da toxina são muito
baixos. O MIP pode tanto ser utilizado em EFS quanto como sensor químico.
Theodoridis e Manesiotis (2002) desenvolveram a síntese de um MIP para
análise de cafeína por polimerização em massa utilizando como monômeros EDMA
e MAA, e AIBN como iniciador em acetonitrila como solvente. Diferentes protocolos
de lavagem e eluição foram desenvolvidos para otimizar a extração de cafeína das
amostras. O MIP sintetizado foi aplicado em cartuchos e utilizado na técnica de
extração em fase sólida. Observou-se que houve boa retenção da cafeína no
cartucho contendo o MIP na etapa de lavagem, com recuperação média de 23%, ao
contrário do cartucho com o NIP (polímero não impresso), onde a retenção não foi
tão eficiente, com recuperação em torno de 76%. O protocolo de extração otimizado
foi utilizado para extração direta da cafeína em bebidas e plasma humano fortificado
com cafeína, com recuperação em torno de 83% nos dois casos.
Theodoridis et al. (2003), utilizaram a técnica do “dummy template” para a
síntese de um MIP seletivo para a escopolamina. Essa técnica envolve a utilização,
como molécula molde, de um análogo estrutural ao analito de interesse, sendo a

31
hiosciamina a molécula selecionada. O MIP foi sintetizado pela técnica de
polimerização em massa, utilizando EDMA, MAA e AIBN em tolueno. Estudos
visando a otimização da EFS para a extração da escopolamina foram realizados, até
se observar que na etapa de lavagem o analito é mais retido em MIP do que em NIP
e o contrário na eluição, onde deve haver maior recuperação em MIP. O MIP
apresentou seletividade para a extração da escopolamina em amostras biológicas,
como urina e soro bovino e humano, entretanto com uma recuperação de 46 a 59%
nas amostras de urina e de até 79% nas amostras de soro.
Scorrano, Longo e Vasapollo (2010) elaboraram a síntese de um MIP para
análise de 1-metiladenosina (1-MA) por polimerização em massa (EDMA, MAA,
AIBN) em acetonitrila:água (4:1) para aplicação na técnica de EFS, superando
dificuldades relacionadas à impressão molecular de substâncias polares, devido a
não solubilidade em solventes orgânicos. A seletividade do MIP foi avaliada
observando-se a recuperação de compostos análogos à 1-MA, como adenosina e
citidina, demonstrando que o MIP foi altamente seletivo para 1-MA. A recuperação
na extração de 1-MA foi de 95% para o MIP, 50% para o cartucho C18 e apenas 36%
para o NIP, mostrando que a retenção foi seletiva. O protocolo de extração adotado
foi eficaz na extração de 1-MA de amostras de urina humana fortificadas,
concentrando a amostra e eliminando interferentes da matriz.
Blomgren e colaboradores (2002) desenvolveram um método para a extração
de clembuterol a partir de amostras de urina bovina usando um polímero de
impressão molecular (MIP). A quantificação de clenbuterol na urina foi realizada
utilizando CLAE com detecção por UV. O MIP foi produzido usando brombuterol
como molécula molde (dummy template) e a seletividade do MIP, para clembuterol,
foi testada contra um polímero não-impresso (NIP). As condições da extração foram
otimizadas de modo a se obter uma maior retenção do analito em MIP do que em
NIP, conseguida na lavagem com acetonitrila-ácido acético (98:2). A retenção de
clembuterol em MIP foi seletiva e melhor avaliada em amostras de urina diluída,
devido a menor força iônica da matriz. Os resultados das análises em CLAE
mostraram que a extração de clembuterol utilizando MIP foi linear no intervalo de 0,5
– 100 ng mL-1, com recuperação absoluta de 75% nesta faixa de concentração.
Um estudo foi desenvolvido para a determinação de cloridrato de tramadol em
fluidos biológicos, utilizando a técnica de MISPE para a limpeza da amostra e
quantificação do analito por CLAE com detecção por UV. Os polímeros impressos

32
(MIPs) foram preparados por polimerização em massa utilizando ácido metacrílico
(MAA) como monômero funcional, dimetacrilato de etilenoglicol como agente de
reticulação e clorofórmio como solvente porogênico. O MIP sintetizado foi usado
como adsorvente em EFS para a extração de cloridrato de tramadol a partir de
plasma e urina humanos. Parâmetros que podem afetar a seletividade do MIP em
relação ao NIP e a eficiência de extração, como a quantidade de MAA, o solvente de
síntese e o pH foram avaliados. A seletividade do MIP foi verificada através da
avaliação da retenção de várias substâncias com estruturas moleculares similares
ao tramadol no MIP, como difenidramina, morfina e dextrometofano. O limite de
quantificação para o cloridrato de tramadol em amostras de urina foi de 3,5 µg L-1 e
de 8,5 µg L-1 para amostras de plasma. As recuperações para amostras de plasma
e urina foram maiores do que 91%.
Gholivand e colaboradores (2010) sintetizaram alguns MIPs para extração em
fase sólida e determinação de furosemida em plasma humano. Uma abordagem
computacional foi utilizada para a prever a melhor interação do analito com o
monômero funcional (MF), com base na comparação da energia de ligação dos
complexos formados entre o analito e os MFs. Alguns MFs foram escolhidos para a
síntese dentre os que apresentaram maior energia de ligação com a furosemida. A
acrilamida foi o MF que produziu o MIP com maior capacidade adsortiva e
seletividade em relação ao NIP, sendo selecionado para a aplicação em EFS. Os
MIPs foram sintetizados por polimerização em massa, com EDMA como agente de
reticulação e acetona como solvente. Os solventes utilizados na EFS foram
otimizados quanto à recuperação da furosemida, nas etapas de carregamento,
lavagem e eluição do analito. A furosemida foi quantificada por CLAE-UV, com
linearidade na faixa de concentração de 0,075 – 3,5 µg mL-1 e limite de quantificação
no plasma de 0,043 µg mL-1.
Embora a maioria dos trabalhos de síntese de MIP sejam por polimerização
em massa, existem muitas abordagens que utilizam a técnica por precipitação,
visando a obtenção de partículas mais uniformes e com maior rendimento de sítios
de reconhecimento, pois a etapa de trituração é abolida.
Kareuhanon e colaboradores (2009) desenvolveram um método de
determinação de nevirapina por MISPE em plasma humano. A síntese do MIP foi
feita pela técnica de polimerização por precipitação, utilizando ácido metacrílico
como MF, trimetacrilato de trimetilolpropano (TRIM) como agente de ligação

33
cruzada, peróxido de benzoíla como iniciador e como solvente uma mistura de
Tetrahidrofurano:metanol:água (THF:MeOH:água 5:4:1). Foram sintetizados 4
polímeros, cada um com uma molécula molde análoga à nevirapina (nicotinamida,
benzamida e benzofenona), além do próprio analito como molde. O MIP com maior
adsorção para a nevirapina foi o sintetizado em nicotinamida, confirmando estudos
de energia de ligação realizados por modelagem molecular pelos autores. A
seletividade entre MIP e NIP foi avaliada em ensaio de adsorção, onde houve uma
maior adsorção em MIP somente em concentrações acima de 0,5 mM e na etapa de
lavagem do cartucho de EFS. Os cartuchos (MIP e NIP) foram carregados com
nevirapina e lavados com 10 alíquotas de 1 mL de acetonitrila. A recuperação de
nevirapina se mostrou elevada no processo de lavagem em MIP (92%) após a quinta
alíquota de lavagem (maior retenção), enquanto em NIP a recuperação foi de 79%
na primeira lavagem. No cromatograma obtido após a extração por MISPE,
observou-se a eliminação de interferentes.
Zhang et al. (2006) utilizaram a técnica de MISPE para análise de Bisfenol A
(BPA), um composto usado na produção de plásticos policarbonatados e resinas
epóxi e considerado um perturbador endócrino. As amostras eram de origem
biológica e ambiental, podendo conter traços de BPA, o que levou à necessidade de
concentração da amostra. Os polímeros foram preparados por polimerização por
precipitação e usados como adsorventes para extração direta de BPA a partir de
diferentes amostras biológicas e ambientais (soro, urina de porco, água da torneira e
camarão). Os monômeros usados foram 4-vinilpiridina (MF) e TRIM (ALC) e o
iniciador foi o AIBN, dissolvidos em uma mistura de acetonitrila e tolueno (ACN:
Tolueno 3:7 v/v). Os MIPs sintetizados variaram somente na quantidade de molde
adicionado ou ausência do mesmo, no caso do NIP. O polímero que apresentou a
maior seletividade e capacidade de adsorção foi o que continha maior concentração
de BPA (8 mmol) na síntese, sendo este utilizado como adsorvente na EFS. O
protocolo de extração foi otimizado e as condições ótimas para condicionamento,
carregamento, lavagem e eluição foram obtidas. Na etapa de lavagem com
acetonitrila, o MIP foi capaz de reter o analito com maior intensidade do que o NIP,
com recuperação de 99 e 33% de BPA, respectivamente. As análises foram
realizadas em um intervalo de concentração de 2 – 20 mM. As recuperações de BPA
para as amostras utilizando MISPE foram 65,8, 82,3, 76,0 e 75,97% para soro
humano fortificado, urina de porco, água da torneira e camarão, respectivamente.

34
Comparado com o cartucho C18, a técnica de MISPE permitiu uma melhor linha de
base, maior eficiência na separação cromatográfica e maior recuperação do analito.
Outros autores sintetizaram MIPs para aplicação em EFS por precipitação,
como Cacho et al. (2003) para extração de triazinas (herbicidas) em amostras de
vegetais. O MIP foi sintetizado em tolueno, com ácido metacrílico e EDMA como
monômeros, AIBN como iniciador e propazina como molécula molde. O
procedimento de MISPE foi otimizado e aplicado à extração das triazinas em batata,
ervilha e amostras de extratos de milho, permitindo a limpeza da amostra. No
entanto, alguns interferentes da matriz, fortemente ligados à matriz polimérica de
forma não específica, dificultaram a quantificação de algumas triazinas. Assim, o
polímero não impresso (NIP) foi utilizado para a remoção destes interferentes, antes
da percolação da amostra em MIP, sendo estes quase completamente removidos,
permitindo a determinação das triazinas em níveis de concentração abaixo do limite
máximo estabelecido para estes resíduos, tornando o procedimento desenvolvido
adequado para o monitoramento desses analitos em amostras de vegetais.
3.2 ESCOLHA DA MOLÉCULA MOLDE
O projeto originou-se inicialmente da necessidade de desenvolver um teste
diagnóstico para intoxicação por neurotoxinas produzidas por microorganismos
aquáticos (cianobactérias), aplicável tanto a análise de água quanto a análise de
material biológico. Apesar de alguns acidentes já bem documentados (AZEVEDO et
al., 2002), este tipo de intoxicação ainda é subnotificada no Brasil por falta de um
teste diagnóstico rápido e simples, que possa substituir os atuais ensaios biológicos
e as análises por CLAE-EM (HUMPAGE; MAGALHAES; FROSCIO, 2010). As cinco
neurotoxinas de maior incidência foram selecionadas e o projeto original visava a
construção de um cartucho de extração em fase sólida composto de polímeros de
impressão molecular que adsorvessem as neurotoxinas. Este cartucho poderia ser
aplicado na triagem de águas de consumo ou de material biológico. A análise
dessas neurotoxinas é dificultada pela natureza química destes compostos,
alcaloides extremamente solúveis em água. A extração seletiva permitiria o emprego
de uma técnica analítica menos seletiva para a quantificação, como a CLAE
convencional para a análise dos resíduos. Essa técnica é mais disponível nos

35
laboratórios do que a CLAE-EM, que seria utilizada, então, como técnica de
confirmação.
Para este trabalho, uma neurotoxina foi selecionada para os primeiros
estudos, a saxitoxina. A saxitoxina (STX) é um composto de elevada polaridade,
produzido pelas cianobactérias. O padrão analítico não é fabricado no Brasil, sendo
de alto custo de aquisição. A viabilidade dessa pesquisa veio da possibilidade de
trabalharmos com padrões secundários, produzidos em berçários de cianobactérias
aqui no Rio de Janeiro. O extrato bruto rico em saxitoxinas utilizado nos primeiros
testes foi fornecido pelo Laboratório de Ecofisiologia e Toxicologia de Cianobactérias
da Universidade Federal do Rio de Janeiro (LECT – IBCCF – UFRJ). No entanto, a
concentração da toxina neste extrato era muito baixa (86 µg/mL) e não foi possível
obter quantidade suficiente para os testes de síntese, ainda que suficientes para a
parte analítica.
A alternativa foi a busca na literatura de moléculas semelhantes, para serem
utilizadas como molécula molde na síntese dos polímeros de impressão molecular,
no lugar da toxina, mais conhecido na literatura de síntese de MIPs por “dummy
template” (KAREUHANON et al., 2009). Para tal foi necessário um levantamento
bibliográfico e um estudo de modelagem molecular para seleção de possíveis
moléculas candidatas, que possuíssem volume molecular igual ou ligeiramente
maior que a toxina e que possuíssem pontos de ancoragem, ou seja, grupos
funcionais doadores e aceptores de ligação de hidrogênio, por exemplo,
espacialmente dispostos ou próximos como na toxina.
Para a realização dos estudos de modelagem molecular, contamos com o
apoio do Grupo de Modelagem Molecular e Estudos ADMETox in silico, grupo
de pesquisa coordenado pela Profa. Dra. Monique Brito, da Faculdade de Farmácia
da UFF, onde essa etapa do estudo foi desenvolvida.
Primeiramente, foi feita uma busca na literatura de compostos com possíveis
características em comum com a saxitoxina, além de busca na base de dados do
SciFinder. Para a seleção dos compostos, selecionaram-se alguns grupos que
contivessem particularidades comuns à saxitoxina, como alcaloides de origem
natural, toxinas, agrotóxicos, agonistas/antagonistas do canal de sódio e
aminoácidos precursores da biossíntese da STX. Alguns parâmetros foram
observados nesses compostos como peso molecular, número de grupos doadores e
aceptores de ligação de hidrogênio (importantes para a interação com o monômero

36
funcional no polímero) e posição destes grupos, além da estrutura em 3D e
facilidade de obtenção.
Os compostos selecionados na busca foram encaminhados para o laboratório
da Profa. Monique, onde foram realizados os estudos de modelagem molecular com
metodologia particular deste e selecionados alguns compostos para a síntese dos
MIPs.
3.2.1 Estudos de modelagem molecular
Foram encontrados na literatura ao todo 40 compostos candidatos
promissores à molécula molde para a STX. Destes, somente 7 foram selecionados
para os estudos de modelagem molecular: Fisostigmina, Colchicina, Papaverina,
Amilorida, Escopolamina, Praziquantel e um glicopiranosil sintético a base de
asparagina e N-acetilglicosamina (Tabela 1).
Tabela 1. Compostos selecionados para os estudos de modelagem
Composto Peso Molecular Grupos doadores de ligação hidrogênio
Grupos aceptores de ligação hidrogênio
Saxitoxina* 287,28 6 10
Fisostigmina 261,32 1 5
Colchicina 385,41 1 6
Papaverina 339,39 5 0
Amilorida 229,63 4 3
Escopolamina 303,35 1 5
Praziquantel 312,41 0 4
Glicopiranosilamina 335,31 7 9
* Colocada na tabela somente para fins comparativos
Os parâmetros calculados através do estudo de modelagem foram o volume
molecular, a área e as distâncias entre os grupos aceptores/doadores de ligação de
idrogênio presentes nas extremidades da molécula. Os resultados obtidos estão na
Figura 9.

37
Figura 9. Compostos candidatos a molécula molde para a STX
As moléculas escolhidas através do estudo de modelagem molecular para a
síntese do MIP como molécula molde para análise de saxitoxina e seus análogos,
foram a colchicina e papaverina , dois alcaloides de origem natural e a amilorida,
um bloqueador de canal de sódio epitelial presente nos túbulos distais renais. Os 3
compostos foram escolhidos principalmente pelo volume molecular, posição dos

38
grupos aceptores e doadores de ligação de hidrogênio e estrutura em 3D
aparentemente semelhante à STX.
Não se obteve resultado satisfatório na síntese de MIPs com a colchicina e
papaverina. A polimerização não ocorreu, provavelmente devido a presença de
grupos metoxi (-OCH3) que funcionaram como inibidores da polimerização,
consumindo o iniciador peróxido de benzoíla, reagente que inicia a reação via
produção de radicais livres. Houve a formação de subprodutos, confirmado através
de cromatografia em camada fina.
A amilorida possui um grupo guanidina também presente na STX. Entretanto,
sugeriu-se que seria necessária uma modificação estrutural na posição 6 do anel
pirazínico ocupado pelo átomo de Cl, para aumentar o volume molecular. Através da
modelagem, foi possível visualizar que o grupo ideal a ser adicionado seria oriundo
de uma substituição nucleofílica do Cl por um derivado ciclohexil ou fenil com a
hidroxila (OH) em posição orto em relação ao grupo nucleófilo, que poderia ser um
grupo amino ou um sal sódico de enxofre (R-NaS), como pode ser visto na Figura
10.
Figura 10. Modificações na estrutura da amilorida sugeridas na modelagem molecular
Escolhemos para fazer a substituição nucleofílica do átomo de Cl da amilorida
o composto p-aminofenol. Contudo, a amilorida é comercializada na forma de sal de
cloridrato, o que impede a solubilidade do composto em solventes orgânicos. Para
solucionar esta questão, partiu-se para a tentativa de extração líquido-líquido do
cloridrato de amilorida para se obter a substância na forma de base livre,
aumentando sua solubilidade em solventes orgânicos. Isto facilitaria tanto a reação

39
de substituição nucleofílica quanto a de síntese do MIP. Entretanto, não obteve-se
sucesso na extração da amilorida como base livre.
Assim, tentou-se conduzir a reação de substituição nucleofílica do átomo de
Cl da amilorida pelo nucleófilo p-aminofenol com o cloridrato de amilorida, segundo
a literatura (TANDON; MAURYA, 2009), utilizando água como solvente e
temperatura da reação a 50ºC, porém sem sucesso. Outras tentativas utilizando
etanol, isopropanol e dimetilsulfóxido (DMSO) como solvente foram feitas, mas
ocorreu a degradação do p-aminofenol. A reação à temperatura ambiente e sob
nitrogênio também não ocorreu.
Um tempo maior seria necessário para o desenvolvimento desta etapa de
síntese, que demonstrou ser mais complexa do que originalmente pensado. Então, a
alternativa foi trabalhar com o cloridrato de amilorida sem modificações estruturais.
Na ausência da saxitoxina, a solução encontrada foi utilizar o próprio fármaco para
os ensaios analíticos afim de comprovar a seletividade dos polímeros sintetizados.
Sendo assim, a síntese dos MIPs foi realizada utilizando o cloridrato de
amilorida como molécula molde e o mesmo analito foi utilizado para os ensaios de
adsorção e extração em fase sólida utilizando os polímeros sintetizados.
3.2.2 Propriedades físico-químicas do cloridrato de amilorida
O cloridrato de amilorida (AMI) é um sal obtido a partir de uma base
moderadamente forte, a amilorida (9, Figura 11). A amilorida possui nomenclatura
IUPAC 3,5-diamino-6-cloro-N-(diaminometileno)-2-pirazinocarboxamida, e consiste
em um anel pirazínico substituído por um grupo acilguanidina na posição 2, dois
grupos amino nas posições 3 e 5 e um átomo de Cl na posição 6. O sal
normalmente apresenta-se cristalino na forma de monocloridrato di-hidratado, de
coloração amarelo-esverdeada e inodoro. A fórmula molecular do composto é
C6H8ClN7O.HCl.2H2O e o peso molecular correspondente é 302,12 (BENOS, 1982;
THAKRAL; MADAN, 2008; MAZZO, 1986).
O grupo guanidínico possui importante papel na molécula de amilorida, pois é
neste grupo que ocorre a protonação, com ressonância da carga entre os
nitrogênios guanidínicos, fazendo com que a amilorida seja considerada uma base
fraca. Todos os hidrogênios da amilorida são intercambiáveis em solução aquosa
(BENOS, 1982).

40
Figura 11. Estrutura da amilorida (9)
A amilorida possui um valor de pKa em torno de 8,70 a 25°C, confirmado
tanto experimentalmente por titulacão aquosa quanto estimado através de cálculos
teóricos como a afinidade por prótons na fase gasosa, a entalpia de solução e
cálculos semiempíricos (BOCK; SCHLEGEL; SMITH, 1981; MAZZO, 1986).
Jozwiakowski e colaboradores (1993) relataram que o cloridrato de amilorida
pode cristalizar na forma de sal di-hidratado, disponível em duas formas polimórficas
A e B, sendo que estas podem ser desidratadas obtendo-se a forma cristalina
anidra. A forma anidra rapidamente se reidrata retornando ao polimorfo A di-
hidratado após exposição à umidade relativa do ambiente. Os polimorfos di-
hidratados A e B tem pontos de fusão, espectros de FTIR e solubilidades
semelhantes. Contudo, a técnica de difração do raio-X pode diferenciá-los e a
quantificação pode ser estimada em amostras com a mistura dos dois polimorfos,
sendo o polimorfo A mais fisicamente estável.
O ponto de fusão do cloridrato de amilorida (AMI) anidro é 293,5°C e da forma
di-hidratada é 288°C, com decomposição. A forma hidratada do sal de AMI pode ser
convertida na forma anidra pela secagem a 100°C a pressão reduzida. O cloridrato
de amilorida é praticamente insolúvel em acetona, clorofórmio, éter etílico e acetato
de etila, livremente solúvel em dimetilsulfóxido, ligeiramente solúvel em isopropanol,
etanol (1,96 mg mL-1) e água, além de moderadamente solúvel em metanol. A
limitada solubilidade de AMI em água é típica de uma base orgânica e aumenta com
a diminuição de pH. Enquanto em pH 4,8 a solubilidade é 5,2 mg mL-1, em pH 10,0
ela reduz para 0,3 mg mL-1 (MAZZO, 1986).
Um estudo de estabilidade de AMI em soluções aquosas foi realizado por Li,
Moore e Tattam (1999). A fotodegradação de AMI foi avaliada em solução aquosa
deaerada a 30 °C na faixa de pH entre 4,5 e 11 através de espectrofotometria e
CLAE em fase reversa. A forma neutra do fármaco presente na solução alcalina se

41
degrada aproximadamente 3 vezes mais rápido do que a forma catiônica. O
processo de fotorreação inicial envolve decloração de AMI e o mecanismo de
fotólise parece envolver a formação de um cátion radicalar que facilita a decloração.
Observou-se que a atividade de fotossensibilização de AMI predominantemente
ocorre na presença de substratos de oxigênio singlete e não por um mecanismo de
formação de radicais livres. No entanto, AMI é um fraco fotossensibilizador, tendo
atividade somente sob condições de radiação intensa, em relação a outros
diuréticos, tais como furosemida e hidroclorotiazida, sob as mesmas condições
experimentais.
O espectro de varredura no ultravioleta de AMI obtido em solução de ácido
clorídrico 0,1 N apresenta máximos de absorção em 212, 285 e 363 nm, sendo que
este último representa seu λmáx. A molécula emite fluorescência a 420 nm, o que
possibilita a sua determinação por técnicas que utilizam este tipo de detecção
(MAZZO, 1986; USP, 1989).
3.2.3 Métodos para determinação de cloridrato de amilorida
Diversos métodos têm sido relatados para determinação de AMI em fluidos
biológicos como urina e plasma e em formulações farmacêuticas, incluindo
espectrofotometria, CLAE, quimioluminescência, polarografia com pulso diferencial,
métodos potenciométricos, entre outros. Embora a determinação por alguns
métodos como CLAE seja altamente sensível, estes requerem a eliminação
preliminar de interferentes e pré-concentração do analito no caso de amostras
biológicas, por extração líquido-líquido, técnica que envolve a utilização de grandes
volumes de solventes orgânicos. Isto acarreta desvantagens como baixa
recuperação do analito, pois os procedimentos de extração se baseiam em reações
de equilíbrio, e aumento do tempo de análise (MIRMOMTAZ; ENSAFI;
SOLEIMANIAN-ZAD, 2009).
Em preparações farmacêuticas, as amostras contendo AMI geralmente não
necessitam de tratamento prévio, pois os excipientes não interferem nas análises
devido a serem insolúveis e assim podem ser eliminados por um processo de
filtração simples (ZECEVIC et al., 2000). No caso de matrizes complexas como
plasma e urina, utilizam-se técnicas preliminares de preparação das amostras para
eliminação de interferentes, como a extração em fase sólida e a extração líquido-

42
líquido previamente à análise por CLAE (FORREST et al., 1988; BI; COOPER;
CÔTÉ, 1992).
A quantificação de AMI por métodos espectrofotométricos são realizados
somente para formulações farmacêuticas, pois apresentam elevado limite de
quantificação e maior sensibilidade aos interferentes (TORAL et al., 2002; ABDEL-
HAY, 2008). Em um estudo de comparação entre os métodos espectrofotométricos
no UV e CLAE em fase reversa com detecção no UV, Kartal e Erk (1999)
observaram uma boa precisão, exatidão e recuperação para ambos os métodos.
Entretanto, o método cromatográfico é mais versátil e mais seletivo para a
determinação de AMI na presença de produtos de degradação e pode ser aplicado a
uma variedade de matrizes.
Halvatzis et al. (1994) relataram um método automatizado de determinação
de AMI por quimioluminescência baseado na oxidação com N-bromosuccinimida em
solução alcalina. O método foi capaz de analisar o composto em formulações
farmacêuticas em uma faixa de 0,5 a 15 µg mL-1 com limite de detecção de 0,16 µg
mL-1, erro relativo de 1,7% e com recuperação média de 95,5% entre as amostras
comerciais analisadas.
Um método simples e rápido para análise de AMI foi desenvolvido utilizando
um sistema de injeção de fluxo monocanal com transdução fluorimétrica, para
amostras de soro humano e formulações farmacêuticas. O AMI era retido
transitoriamente em uma coluna de troca catiônica de gel Sephadex SP-C25
colocada na área de detecção dentro da célula. A determinação de AMI foi realizada
sem nenhuma reação de derivatização, medindo diretamente a fluorescência
intrínseca do analito, com limite de detecção de 0,92 e 0,33 mg L-1 para volumes de
injeção de 100 e 600 µL, respectivamente. As recuperações de AMI em amostras de
soro fortificadas e em formulações foram de 96,3 a 105% e 98 a 100,6%,
respectivamente (DOMINGUEZ-VIDAL; ORTEGA-BARRALES; MOLINA-DIAZ,
2002).
Martín et al. (1999) utilizaram o método de polarografia de pulso diferencial
para a quantificação simultânea de AMI e HCTZ em formulações farmacêuticas. Os
resultados obtidos foram de acordo com o conteúdo declarado pelos fabricantes dos
comprimidos contendo AMI e HCTZ analisados, sendo estes comparados com a
quanficação por CLAE. Não houve diferença significativa entre os dois métodos
utilizados para quantificação e a diferença entre as duas marcas analisadas foi

43
menor do que 15%, aceito pela legislação. O desvio padrão relativo calculado foi de
0,5 a 3,4% (n=6) entre as amostras quantificadas em dias diferentes.
Ensafi e Allafchian (2008) desenvolveram um novo sensor potenciométrico a
base de membrana de Policloreto de vinila (PVC) para determinação seletiva de AMI
em comprimidos e urina. O método é baseado na formação de um par iônico entre a
amilorida e tetrafenilftalato de sódio como material eletroativo e dibutilftalato (DBP)
como íon exclusor. O eletrodo indicador (amilorida) foi montado com uma solução
interna de AMI e na ponta colocado uma membrana composta do par iônico AMI-
tetrafenilftalato de sódio além de DBP e PVC. Um eletrodo de calomelano foi usado
como referência. O sensor de membrana apresentou resposta satisfatória e rápida
para a amilorida em um intervalo de concentração de 1,0 × 10-2 a 1,0 × 10-6 mol L-1
com um limite de detecção de 9,9 × 10-7 mol L-1 e exibiu alta seletividade para
amilorida frente a um grande número de compostos relacionados. Os mesmos
autores também desenvolveram um outro eletrodo semelhante feito de membrana
polimérica, mas utilizando ds-DNA de esperma de salmão-amilorida como par iônico
e nitrofeniloctiléter como um íon exclusor para quantificação de amilorida em
formulações, plasma e urina (ALLAFCHIAN; ENSAFI, 2010).
Em outro método potenciométrico desenvolvido para quantificação de AMI em
comprimidos e urina, um eletrodo de grafite foi usado como suporte para imobilizar a
membrana de ds-DNA-amilorida. A técnica de voltametria por pulso diferencial foi
usada para determinar a mudança da intensidade do sinal de oxidação das bases
guanina e adenina antes e depois da interação com a amilorida. A diminuição na
intensidade do sinal se mostrou proporcional para uma faixa de concentração de
amilorida de 0,75 a 240 µM, com um limite de detecção de 0,5 µM, com resultados
satisfatórios (MIRMOMTAZ; ENSAFI; SOLEIMANIAN-ZAD, 2009).
A determinação simultânea de AMI e atenolol (ATE), propranolol (PRO) e
dipiridamol (DIP) por espectrometria de fluorescência foi desenvolvida por Pulgarín,
Molina e López (1998), em amostras farmacêuticas comerciais. Para aumentar a
seletividade do método um espectrômetro de fluorescência com ângulo variável
sincronizado foi utilizado, de modo que o espectro foi obtido pela varredura de
comprimentos de onda de excitação e emissão a diferentes velocidades, sendo a
diferença entre eles não constante. A combinação da espectrometria de varredura
sincronizada com técnicas derivativas aumentou a sensibilidade do método em
comparação ao espectro convencional, devido a amplitude do sinal ser inversamente

44
proporcional à largura da banda do espectro original. O método foi adequado para a
determinação dos analitos em uma faixa de concentração na ordem de ng mL-1, com
limites de detecção de 5,2, 1,9, 2,1 e 0,1 ng mL-1 para ATE, PRO, AMI e DIP,
respectivamente.
Um método para a estimativa simultânea de AMI e HCTZ em plasma humano
por CLAE-EM/EM em fase reversa foi validado utilizando triantereno e
hidroclorotiazida13C,d2 como padrão interno. Os analitos e padrões internos foram
extraídos do plasma através de extração em fase sólida simples. O limite de
quantificação do método foi de 0,1 e 5 ng mL-1 e a recuperação média foi de 41,1 e
81,5% para amilorida e hidroclorotiazida, respectivamente. O coeficiente de variação
do ensaio foi de menos de 11,2% e 5,2% e a precisão foi de 89,0 – 98,1 e 96,6 –
102,9% para AMI e HCTZ, respectivamente. Os autores concluíram que o método
pode ser aplicado a estudos farmacocinéticos envolvendo os dois fármacos
(JANGID; TALE; VAIDYA, 2011).
Um grupo de pesquisadores recentemente utilizou o método fluorimétrico para
determinação de AMI e furosemida (FUR) em formulações farmacêuticas
(PERALTA; FERNÁNDEZ; MASI, 2010) e em urina (PERALTA; FERNÁNDEZ;
MASI, 2011). Nos dois casos, as amostras foram tratadas em uma membrana de
nylon composta de poliamida de 0,45 µm antes da análise, devido a determinação
direta convencional promover a sobreposição do espectro de fluorescência dos dois
analitos e no caso da urina, também devido à fluorescência de alguns componentes
da matriz. A solução de AMI foi alcalinizada a pH 11 para possibilitar a retenção do
composto na membrana de poliamida (não polar), sendo que a FUR está
predominantemente na forma carregada em pH acima de 5 e não fica retida na
membrana, separando os dois diuréticos. A AMI foi determinada diretamente na
membrana frente a um branco de matriz, ou seja, a urina sem os diuréticos foi
filtrada na membrana e verificou-se que não havia fluorescência. A FUR foi
determinada na solução aquosa, após o pH ser ajustado para 2,7, onde a urina não
fluoresce. O limite de detecção para amostras de urina foi de 0,11 ng mL-1 para AMI
e de 0,35 ng mL-1 para FUR, mostrando a sensibilidade do método, que apresentou
uma recuperação variando de 91,5 a 109,5%.

4 MATERIAIS E MÉTODOS
4.1 EQUIPAMENTOS
A síntese dos MIP e NIP foi conduzida no Laboratório de Química Medicinal
(LQMed) da Faculdade de Farmácia da UFF, sob orientação da profa. Estela Muri.
As moléculas molde e o peróxido de benzoíla usados para a síntese foram
pesados em balança analítica de precisão Bioprecisa JA3003N (Brasil) e o
aquecimento e agitação da reação foram realizados em agitador magnético com
aquecimento Ika RH Basic 2S1 (Alemanha). Os polímeros foram secos à
temperatura ambiente e o solvente residual foi evaporado com auxílio de uma
bomba de alto vácuo Edwards RV3 (Inglaterra) até peso constante.
A tamização dos polímeros, o ensaio de adsorção e a extração em fase sólida
(EFS) foram conduzidos no Laboratório de Toxicologia Analítica (ToxFree) da
Faculdade de Farmácia da UFF, sob orientação da profa. Eliani Spinelli.
Os polímeros foram tamizados em peneiras para análise granulométrica
Granutest (São Paulo, Brasil) de 25 a 62 µm. Foram desprezadas as partículas com
menos de 25 µm e com mais de 62 µm.
Os polímeros e o AMI foram cuidadosamente pesados em balança analítica
de precisão Shimadzu AUW220D (Japão) para a condução dos ensaios de adsorção
e para preparação dos cartuchos de EFS.
Os microtubos, da Eppendorf (Alemanha) utilizados nos ensaios de adsorção
dos polímeros foram colocados em agitação utilizando o homogeneizador de tubos
Phoenix AP 22 (São Paulo, Brasil).
Após o ensaio de adsorção, os microtubos foram centrifugados em uma
centrífuga para microtubos Minispin®, da Eppendorf (Alemanha).

46
Os reagentes para o preparo da solução tampão citrato-acetato pH 6,5 (0,12M
e 0,66 M) utilizada nos ensaios de adsorção foram pesados em balança Gehaka
BG400 (São Paulo, Brasil) e o ajuste do pH foi feito em medidor de pH Tekna T-
1000 (Brasil).
A água ultra-pura foi obtida através de um sistema de purificação de água
Simplicity UV, da Millipore (França).
As soluções de cloridrato de amilorida (AMI), molécula molde escolhida para
os ensaios de adsorção e para a EFS, foram preparadas em água ultra-pura,
solubilizando o sal (cloridrato) com auxílio de um banho ultrassônico, Unique
Ultracleaner 1600A (São Paulo, Brasil). As alíquotas das soluções utilizadas foram
pipetadas com o auxílio de micropipetas monocanal de volume variável P100,
P1000, P5000, Research Plus® da Eppendorf (Alemanha) e P200 da Labnet (EUA).
A EFS foi conduzida em uma câmara de vácuo Supelco VisiprepTM (EUA) com
capacidade para 12 cartuchos acoplado a uma fonte de vácuo. Os polímeros foram
empacotados em cartuchos de polipropileno vazios de 3 mL para EFS, Supelco
(EUA), entre dois “frits” (Supelco, EUA) de polipropileno com porosidade de 20 µm.
Os “frits” foram colocados no cartucho por um dispositivo de inserção de “frits”, da
Supelco (EUA). Os polímeros foram pesados diretamente nos cartuchos.
As leituras das absorbâncias das soluções de AMI obtidas nos ensaios de
adsorção e EFS foram realizadas em espectrofotômetro UV-Visível, Bel Photonics
Spectrophotometer SP 2000 UV (Itália).
Após a EFS, a evaporação do solvente extrator foi feita a 50ºC sob fluxo de
nitrogênio, em um termoreator para tubos (Dry Block), da Marconi (Brasil) provido de
linha de distribuição de nitrogênio.
Os polímeros foram visualizados em microscópio eletrônico de transmissão
FEI Morgagni 268D (EUA), gentilmente cedido pelo Laboratório de Microscopia do
prof. Venício Veiga no Instituto de Microbiologia da UFRJ.
As análises de caracterização dos polímeros por Espectrofotometria no
Infravermelho por transformada de Fourier (FTIR) foram realizadas no Instituto de
Química da UFF, utilizando o equipamento Varian 660-IR FT-IR Spectrometer
(Alemanha) com aplicação direta da amostra.

47
4.2 REAGENTES
Na síntese de MIP e NIP foram utilizados os monômetros ácido metacrílico
(MAA, 99%), dimetacrilato de etilenoglicol (EDMA, 98%) e trimetacrilato de
trimetilolpropano (TRIM, grau técnico) provenientes da Sigma Aldrich (Alemanha). O
peróxido de benzoíla P.S. utilizado foi o da Vetec (Brasil). Os monômeros foram
utilizados sem purificação prévia.
O cloridrato de amilorida (AMI) utilizado na síntese e demais ensaios foi
adquirido na DEG Importação de Produtos Químicos Ltda. (Brasil), com certificado
de análise do fabricante cumprindo as especificações das Farmacopéias Britânica e
Européia (Anexo 8.5).
Os solventes orgânicos usados foram tetrahidrofurano (grau espectroscópico
HPLC/UV), acetonitrila (grau espectroscópico HPLC/UV) e ácido acético glacial
ACS, todos obtidos da Tedia (EUA). O metanol (grau espectroscópico HPLC/UV),
ácido trifluoroacético P.S. e ácido clorídrico P.A. utilizados foram provenientes da
Vetec (Brasil).
Os reagentes usados para fazer a solução tampão citrato-acetato foram
hidróxido de sódio P.A. Vetec (Brasil), ácido cítrico anidro ACS Spectrum (EUA) e
acetato de sódio tri-hidratado USP Sigma Aldrich (Alemanha).
4.3 PROCEDIMENTOS
4.3.1 Síntese dos MIPs e NIPs em cloridrato de amilorida
Os reagentes e solventes foram adicionados nas quantidades listadas na
Tabela 2. Em um balão de fundo redondo bitubulado de 100 mL conectado a um
condensador de refluxo, foi colocado o solvente e, no caso dos MIPs, adicionou-se o
cloridrato de amilorida (AMI). Após a solubilização do composto, dissolveu-se o
ácido metacrílico (MAA) deixando em agitação por 10 min. Posteriormente foram
adicionados o agente de reticulação (EDMA ou TRIM) e o peróxido de benzoíla (PB)
72% (iniciador radicalar). Após a adição dos reagentes, o balão foi fechado com
septo de borracha, a atmosfera dentro do balão foi saturada com gás nitrogênio e a
solução foi borbulhada com o mesmo gás por 10 minutos através de uma cânula de
inox. O balão então foi colocado em banho de óleo, deixando a reação a 60°C por

48
24h. Os NIPs foram sintetizados com o mesmo procedimento, entretanto sem a
adição de AMI.
Tabela 2. Quantidades dos reagentes na síntese dos MIPs e NIPs de cloridrato de amilorida
MIP/NIPAMI
(mmol)*2
MAA
(mmol)
Agente de
Reticulação
(mmol)
PB*4
(mmol)Solvente
1* 0,25 1 1 (TRIM) 0,125 15 mL THF:MeOH:água (5:4:1)
2 0,25 1 2 (TRIM) 0,125 20 mL THF:MeOH:água (5:4:1)
3 0,25 1 3 (EDMA) 0,31 20 mL THF:MeOH:água (5:4:1)
4 0,25 1 2 (TRIM) 0,125 20 mL THF:MeOH (4:1)
5 0,25 1 3 (EDMA) 0,31 20 mL THF:MeOH (4:1)
6 0,25 1 2 (TRIM) 0,125 30 mL THF:MeOH:água (5:4:1)
7*3 0,25 1 5 (EDMA) 0,60 20 mL THF:MeOH:água (5:4:1)
8 0,25 2 5 (EDMA) 0,60 20 mL THF:MeOH:água (5:4:1)
9 0,25 1,5 5 (EDMA) 0,51 20 mL THF:MeOH:água (5:4:1)
10 0,25 2 10 (EDMA) 0,97 20 mL THF:MeOH:água (5:4:1)
11 0,25 1 2,5 (TRIM) 0,30 20 mL THF:MeOH:água (5:4:1)
12 0,25 2 2,5 (TRIM) 0,34 20 mL THF:MeOH:água (5:4:1)
13 0,25 2 1,25 (TRIM) 0,21 20 mL THF:MeOH:água (5:4:1)
* proporção usada segundo Kareuhanon et al. (2009).
*2 ausente em NIP
*3 proporção geral encontrada na literatura
*4 ajustado de acordo com a quantidade de monômero e observações das reações de polimerização
O final da reação foi verificado através da formação de um precipitado branco
leitoso, confirmando a precipitação do polímero. O polímero precipitado foi
transferido com auxílio de metanol para um cartucho de celulose para extrator
soxhlet de 33 x 80 mm, sendo este colocado em um conjunto extrator soxhlet de 250
mL completo (balão de fundo chato, extrator soxhlet e condensador de bolas). O
excesso de reagentes e molécula molde foi extraído por lavagem no extrator soxhlet
com metanol:ácido acético 9:1 (v/v) por algumas horas. O polímero então foi lavado
com metanol ainda no soxhlet para retirar o ácido e, após a lavagem, o cartucho foi
deixado para secagem em temperatura ambiente para eliminação do excesso de
solvente. O polímero foi então transferido para um balão de fundo redondo e
colocado em bomba de alto vácuo até peso constante.

49
Após secos, os polímeros foram macerados levemente em gral e pistilo para
dispersão dos grânulos. Os polímeros MIPs e NIPs preparados foram peneirados em
tamizes de 0,025 a 0,062 mm para a obtenção de partículas entre 25 e 60 µm para o
ensaio de adsorção e EFS.
Após tamizados, os polímeros foram caracterizados quimicamente através de
Espectroscopia no Infravermelho com Transformada de Fourier (FTIR), de acordo
com o item 4.3.2 e morfologicamente por Microscopia Eletrônica de Transmissão
(MET), de acordo com o item 4.3.3 (somente para os polímeros MIP/NIP 12).
4.3.2 Caracterização química por Espectroscopia no Infravermelho com
Transformada de Fourier (FTIR)
Os polímeros MIPs/NIPs 7, 8, 10, 11 e 12, os monômeros MAA, EDMA e
TRIM, além do cloridrato de amilorida foram enviados para o laboratório de
Espectrofotometria em Infravermelho no Instituto de Química da UFF, Campus
Valonguinho. Os espectros foram feitos com aplicação direta da amostra no
equipamento.
4.3.3 Microscopia Eletrônica de Transmissão (MET) dos polímeros MIP/NIP 12
Os polímeros foram suspensos em água ultra-pura na concentração de 1 mg
mL-1 e diluídos em água na concentração de 1:10 em microtubos. As amostras foram
levadas para o Laboratório de Microscopia do prof. Venício Veiga na UFRJ, onde
foram depositadas em um suporte de cobre para visualização direta das amostras
por MET.
4.3.4 Preparo da solução tampão citrato-acetato pH 6,5 para os ensaios de
adsorção e EFS
A solução tampão citrato-acetato pH 6,5 foi preparada pesando-se 23 g de
ácido cítrico anidro (0,12M), 45 g de acetato de sódio tri-hidratado (0,66 M) e 10 g de
hidróxido de sódio. Os sais foram dissolvidos em água ultra-pura, completando-se o
volume a 1000 mL em balão volumétrico. O pH foi ajustado a 6,5 com solução de
NaOH a 70% (p/V) em medidor de pH de bancada.

50
4.3.5 Preparo da solução estoque de AMI a 1 mg/mL
A solução estoque foi preparada pesando-se exatamente cerca de 10 mg de
AMI em balão volumétrico de 10 mL. O sal foi solubilizado em água ultra-pura com
auxílio de banho ultrassônico e a solução foi avolumada. A cada dia de experimento
foi preparada uma nova solução estoque e a partir desta foram retiradas alíquotas
para diluição e obtenção das demais concentrações utilizadas nos ensaios.
4.3.6 Espectro de varredura de AMI em HCl 0,1N e tampão citrato-acetato pH 6,5
O espectro de varredura da solução de AMI foi obtido no espectrofotômetro
UV-Visível na faixa de comprimento de onda entre 260 e 390 nm, utilizando solução
de AMI na concentração de 9 µg mL-1 diluída em tampão citrato-acetato. Os λmáx
foram comparados aos obtidos no espectro de varredura feito em HCl 0,1N na
mesma faixa de comprimento de onda e mesma concentração. O HCl 0,1N é o
solvente preconizado pela Farmacopéia americana USP XXII (1989) para análise de
cloridrato de amilorida.
4.3.7 Preparo da curva de calibração em solução tampão citrato-acetato pH 6,5
A curva de calibração para o ensaio de adsorção foi feita a partir da solução
estoque de AMI a 1 mg/mL em água ultra-pura. As diluições foram preparadas em
triplicata, avolumando-se com tampão citrato-acetato pH 6,5, de forma a obter a
curva em uma faixa de concentração de 0,1 a 20 µg/mL. As leituras das
absorbâncias foram feitas em espectrofotômetro UV-Vis a 363 nm (λmáx).
4.3.8 Ensaio de adsorção variando a quantidade de polímero
Para a realização deste ensaio, foram escolhidos os polímeros MIPs/NIPs 10,
11 e 12 pela maior quantidade de material obtido. Pesou-se 7,5, 15 e 30 mg em
duplicata de cada MIP e NIP em microtubos de 2,0 mL. Adicionou-se 1,5 mL de
solução de cloridrato de amilorida em tampão citrato-acetato pH 6,5 a 9 µg mL-1 em
cada tubo, preparadas a partir da solução estoque a 1 mg/mL. Os tubos foram
colocados no agitador de tubos por inversão por 16 horas e centrifugados a 10000

51
rpm por 10 min. O sobrenadante foi recolhido e levado para leitura em
espectrofotômetro UV-Vis a 363 nm. Solução-controle, sem polímero, foi submetida
às mesmas condições do ensaio.
4.3.9 Ensaio de adsorção de AMI em MIP/NIP 7, 8, 10 e 11 variando a
concentração de AMI
O ensaio de adsorção de cloridrato de amilorida nos polímeros em solução de
tampão citrato-acetato pH 6,5 foi conduzido seguindo a metodologia adaptada de
Kareuhanon et al. (2009) e Chen, Chen e Lin (2001).
Para a realização deste ensaio, foram escolhidos os polímeros MIPs/NIPs 7,
8, 10 e 11, . Pesou-se, em triplicata, 7,5 mg de cada MIP e NIP em microtubos para
centrífuga de 2,0 mL. Uma alíquota de 1,5 mL de solução de cloridrato de amilorida
em tampão citrato-acetato pH 6,5 nas concentrações de 3, 6, 9, 12 e 15 µg mL-1 foi
adicionada em cada tubo. As soluções foram diluídas e preparadas a partir da
solução estoque a 1 mg/mL. Os tubos foram colocados no agitador de tubos por
inversão por 16 horas e centrifugados a 10000 rpm por 10 min. O sobrenadante foi
recolhido e levado para leitura em espectrofotômetro UV-Vis a 363 nm. Soluções-
controle, sem polímero, em cada concentração foram submetidas às mesmas
condições do ensaio.
4.3.10 Ensaio de adsorção de AMI em MIP/NIP 10 e 12 variando a concentração de
AMI
Pesou-se 7,5 mg em duplicata de cada MIP e NIP em microtubos para
centrífuga de 2,0 mL. Adicionou-se 1,5 mL de soluções de cloridrato de amilorida em
tampão citrato-acetato pH 6,5 na faixa de concentração de 6 a 75 µg mL-1 para
MIP/NIP 10 e de 6 a 60 µg mL-1 para MIP/NIP 12 em cada tubo, preparadas a partir
da solução estoque a 1 mg/mL. Os tubos foram colocados no agitador de tubos por
inversão por 16 horas e centrifugados a 10000 rpm por 10 min. O sobrenadante foi
recolhido e levado para leitura em espectrofotômetro UV-Vis a 363 nm. Soluções-
controle, sem polímero, em cada concentração foram submetidas às mesmas
condições do ensaio.

52
4.3.11 Ensaio de adsorção de AMI nos polímeros em THF: MeOH: água (5:4:1)
Neste ensaio foram testados os polímeros MIPs/NIPs 7, 8, 10, 11 e 12.
Pesou-se 7,5 mg em duplicata de cada MIP e NIP em microtubos de 2,0 mL.
Adicionou-se 1,5 mL de solução de cloridrato de amilorida em THF: MeOH: água
(5:4:1) a 9 µg mL-1 em cada tubo, preparadas a partir da solução estoque a 1 mg/mL.
Os tubos foram colocados no agitador de tubos por inversão por 16 horas e
centrifugados a 10000 rpm por 10 min. Uma alíquota de 1 mL do sobrenadante foi
recolhida em tubos de vidro e o solvente foi evaporado em um termoreator Dry Block
a 70°C sob nitrogênio. Os resíduos foram solubilizados em 1 mL de tampão citrato-
acetato e levados para leitura em espectrofotômetro UV-Vis a 363 nm. Solução-
controle, sem polímero, foi submetida às mesmas condições do ensaio.
4.3.12 Confecção dos cartuchos para a EFS
O polímero escolhido para a confecção dos cartuchos de EFS foi MIP/NIP 12.
Em um cartucho de polipropileno vazio, com capacidade de 3 mL, foi colocado um
“frit” de 20 µm. Foram pesados 20 mg de MIP/NIP 12 dentro do cartucho e colocou-
se outro “frit” de modo a acondicionar os polímeros entre os “frits”. Os cartuchos
foram cobertos com papel alumínio e guardados à temperatura ambiente até o
momento dos ensaios de extração em fase sólida com impressão molecular
(MISPE).
4.3.13 Procedimento da técnica de MISPE com solução de AMI
Os cartuchos preparados foram colocados em uma câmara de vácuo com
entradas para os cartuchos de EFS, com capacidade para 12 cartuchos. Os
cartuchos foram condicionados aplicando-se 1 mL de metanol, seguido por 1 mL de
água e 1 mL de tampão citrato-acetato, segundo metodologia de Blomgren et al.
(2002).
O ensaio de recuperação do MISPE foi realizado em duplicata para MIP e
NIP. Cada cartucho, após condicionamento, foi carregado com 2 mL de solução de
cloridrato de amilorida a 10 µg mL-1 (20 µg) diluída em tampão citrato-acetato pH 6,5
a partir da solução estoque a 1 mg/mL, aplicando-se o vácuo mínimo necessário

53
para o gotejamento de 1 – 2 gotas por segundo. O eluato foi recolhido para leitura no
espectrofotômetro a fim de avaliar a retenção nessa velocidade de eluição, típica
das aplicações de EFS.
A seguir, em cada cartucho de MIP e NIP, foram aplicados 2 mL de cada
solvente ou mistura solvente testada quanto à capacidade de recuperar a amilorida
retida. Foram testados os seguintes solventes e misturas: metanol, metanol:ácido
trifluoroacético (TFA) a 0,1%, 1%, 2%, 3%, 4% e 5% e metanol:ácido acético a 1%.
Esses solventes foram recolhidos em tubos de vidro e evaporados a 50°C sob
nitrogênio. Os resíduos foram diluídos em 2 mL de tampão citrato-acetato pH 6,5 e
levados para leitura em espectrofotômetro UV-Vis a 363 nm. Uma solução-controle
de amilorida a 10 µg mL-1 foi analisada no espectrofotômetro.
Para avaliação do efeito de um solvente aprótico na técnica de MISPE, foi
feito um teste com eluição em acetonitrila (ACN) e acetonitrila:ácido acético a 1% e
2%. Para isso, os cartuchos (duplicata para MIP e NIP) foram condicionados com 1
mL de metanol:TFA a 2%, 1 mL de água ultra-pura e 1 mL de acetonitrila. O
cartucho foi então carregado com 1 mL de solução de AMI a 20 µg mL-1 (20 µg)
preparada diluindo-se uma alíquota da solução estoque a 1 mg/mL em água ultra-
pura com ACN. A seguir foram aplicados 2 mL do solvente de eluição,
acetonitrila:ácido acético a 1%. Esses solventes foram recolhidos em tubos de vidro
e evaporados a 50°C sob nitrogênio. Os resíduos foram diluídos em 2 mL de tampão
citrato-acetato pH 6,5 e levados para leitura em espectrofotômetro, junto com a
solução-controle de amilorida a 20 µg mL-1.
Para cada ensaio de EFS foi utilizado um novo cartucho. Os cartuchos não
foram reutilizados após os ensaios, sendo descartados após os mesmos, conforme
recomendado por Blomgren et al. (2002).

5 RESULTADOS E DISCUSSÃO
5.1 SÍNTESE DOS MIPs E NIPs
As sínteses dos MIPs e NIPs de cloridrato de amilorida (AMI) foram
conduzidas variando-se a proporção de reagentes (monômero funcional, agente de
reticulação e iniciador) e o volume e tipo de solvente, para ajuste das condições
ideais de polimerização. Estes ajustes foram importantes para a obtenção de
polímeros com características satisfatórias para a EFS. A quantidade de iniciador
radicalar, peróxido de benzoíla, também foi ajustado a cada reação de modo a obter
uma polimerização eficiente, observada pela formação do precipitado branco leitoso
característico do polímero.
Foram obtidos 25 polímeros, sendo 13 MIPs (de 1 a 13) e 12 NIPs (de 2 a
13). Os primeiros polímeros obtidos (MIPs/NIPs 1 a 6) foram considerados
polímeros teste para ajuste da síntese, não sendo utilizados nos testes de adsorção
e EFS. Os MIPs e NIPs numerados de 1 a 3 e de 6 a 13 foram sintetizados em
THF:MeOH:água (5:4:1), enquanto os MIPs e NIPs 4 e 5 o foram em THF:MeOH
(4:1). A mistura de solventes THF:MeOH (4:1) também foi testada para avaliar qual
o efeito da remoção da água na síntese dos polímeros. O MIP 1 e os MIPs/NIPs 2,
6, 11, 12 e 13 contém TRIM como agente de reticulação e os MIPs/NIPs 3, 7, 8, 9 e
10 contém EDMA. Os rendimentos das reações de síntese dos polímeros foram
calculados com base na massa inicial de monômeros e final de polímero obtida, de
acordo com a Tabela 3. O cálculo do rendimento foi feito antes da tamização dos
polímeros. Para polímeros de adição, assumindo-se conversão total, o peso de
polímero formado é igual ao peso de monômero adicionado (CANEVAROLO Jr.,
2006).

55
Tabela 3. Rendimentos das reações de síntese dos MIPs e NIPs
MIP/NIP Proporção de
reagentes*
Volume de
Solvente*3
Massa inicial de
monômeros (g)
Massa final de
polímero (g)
Rendimento
(%)
MIP 1 1:4:4 (TRIM) 15 mL 15 mL (a) 0,427 0,081 19
MIP 2 1:4:8 (TRIM) 20 mL 20 mL (a) 0,765 0,874 114
NIP 2 0:4:8 (TRIM) 20 mL 20 mL (a) 0,765 1,009 131
MIP 3 1:4:12 (EDMA) 20 mL 20 mL (a) 0,854 0,871 102
NIP 3 0:4:12 (EDMA) 20 mL 20 mL (a) 0,854 1,173 137
MIP 4 1:4:8 (TRIM) 20 mL 20 mL (b) 1,532 gelificou -
NIP 4 0:4:8 (TRIM) 20 mL 20 mL (b) 1,532 gelificou -
MIP 5 1:4:12 (EDMA) 20 mL 20 mL (b) 1,708 gelificou -
NIP 5 1:4:12 (EDMA) 20 mL 20 mL (b) 1,708 gelificou -
MIP 6 1:4:8 (TRIM) 30 mL 20 mL (a) 1,532 1,662 108
NIP 6 0:4:8 (TRIM) 30 mL 30 mL (a) 1,532 1,501 98
MIP 7 1:4:20 (EDMA) 20 mL 30 mL (a) 1,349 1,258 93
NIP 7 0:4:20 (EDMA) 20 mL 20 mL (a) 1,349 1,711 127
MIP 8 1:8:20 (EDMA) 20 mL 20 mL (a) 1,436 0,845*2 59*2
NIP 8 0:8:20 (EDMA) 20 mL 20 mL (a) 1,436 1,901 132
MIP 9 1:6:20 (EDMA) 20 mL 20 mL (a) 1,390 1,121 81
NIP 9 0:6:20 (EDMA) 20 mL 20 mL (a) 1,390 1,825 131
MIP 10 1:8:40 (EDMA) 20 mL 20 mL (a) 2,700 3,357 124
NIP 10 0:8:40 (EDMA) 20 mL 20 mL (a) 2,700 3,346 124
MIP 11 1:4:10 (TRIM) 20 mL 20 mL (a) 0,935 0,970 104
NIP 11 0:4:10 (TRIM) 20 mL 20 mL (a) 0,935 0,900 96
MIP 12 1:8:10 (TRIM) 20 mL 20 mL (a) 1,023 1,199 117
NIP 12 0:8:10 (TRIM) 20 mL 20 mL (a) 1,023 1,317 129
MIP 13 1:8:5 (TRIM) 20 mL 20 mL (a) 0,599 0,672 112
NIP 13 0:8:5 (TRIM) 20 mL 20 mL (a) 0,599 0,681 114
* molécula molde: monômero funcional: agente de reticulação (TRIM ou EDMA)
*2 Um pouco de polímero foi perdido no manuseio antes da pesagem
*3 (a) THF:MeOH:água (5:4:1) (b) THF:MeOH (4:1)
O MIP 1 (1:4:4) foi sintetizado segundo as proporções e condições descritas
em Kareuhanon et al. (2009), entretanto obteve-se baixo rendimento (19%),
alterando-se a proporção para (1:4:8), dando origem a MIP/NIP 2, com rendimento
satisfatório. Os MIP/NIP 3 foram sintetizados com EDMA no lugar de TRIM e
proporção de (1:4:12), também com elevados rendimentos. Os rendimentos acima
de 100% podem ser devidos a presença de moléculas de água aderidas ao polímero
e ainda pelas altas quantidades de agente de reticulação utilizada na síntese.

56
Os aspectos dos polímeros obtidos estão descritos na Tabela 4. A troca pela
mistura de solventes para THF:MeOH (4:1) foi com intuito de remover a água do
sistema, sendo obtidos os polímeros MIP/NIP 4 e 5. Os polímeros obtidos não
apresentaram aspecto satisfatório para aplicação em EFS, pois formou-se um
polímero em estado de gel. O processo de gelificação do polímero provavelmente foi
devido à solubilização do polímero nessa mistura de solventes. O polímero sólido
em contato com o solvente tende a inchar através da difusão das moléculas do
solvente para dentro da massa polimérica, formando um gel inchado
(CANEVAROLO Jr., 2006). Após o processo de secagem, estes polímeros
resultaram em partículas muito heterogêneas. Nos polímeros com EDMA (MIP/NIP
5), o processo de gelificação foi mais expressivo, devido à maior massa de
monômeros na reação e o polímero seco apresentou aspecto mais endurecido e de
aparência acrílica. A mistura de solventes então não foi mais utilizada ao longo do
estudo, e portanto o rendimento desses polímeros não consta na Tabela 3.
Tabela 4. Aspecto dos polímeros obtidos
MIP/NIPProporção
AMI*:MAA:ALCSolvente*3 Aspecto do polímero
MIP NIP
1* 1:4:4 (TRIM) 15 mL (a) Baixo rendimento -
2 1:4:8 (TRIM) 20 mL (a) Bom rendimento Bom rendimento
3 1:4:12 (EDMA) 20 mL (a) Bom rendimento Bom rendimento
4 1:4:8 (TRIM) 20 mL (b) gelificação gelificação
5 1:4:12 (EDMA) 20 mL (b) gelificação gelificação
6 1:4:8 (TRIM) 30 mL (a) Partículas < 25 µm Partículas < 25 µm
7 1:4:20 (EDMA) 20 mL (a) Homogêneas (32 – 62 µm) Homogêneas (32 – 62 µm)
8 1:8:20 (EDMA) 20 mL (a) Homogêneas (32 – 62 µm) Homogêneas (32 – 62 µm)
9 1:6:20 (EDMA) 20 mL (a) Heterogêneas Heterogêneas
10 1:8:40 (EDMA) 20 mL (a) Polímero rígido Polímero rígido
11 1:4:10 (TRIM) 20 mL (a) Homogêneas (25 – 62 µm) Homogêneas (25 – 62 µm)
12 1:8:10 (TRIM) 20 mL (a) Homogêneas (25 – 62 µm) Homogêneas (25 – 62 µm)
13 1:8:5 (TRIM) 20 mL (a) Heterogêneas Heterogêneas
* proporção usada segundo Kareuhanon et al. (2009)
*2 ausente em NIP
*3 (a) THF:MeOH:água (5:4:1) (b) THF:MeOH (4:1)

57
Para avaliarmos a possibilidade de produzir uma quantidade maior de
polímeros a cada síntese, MIP/NIP 6 foi sintetizado, onde todos os reagentes foram
dobrados, mantendo-se a proporção (1:4:8) usada em MIP/NIP 2. No entanto, o
volume do solvente foi aumentado para 30 mL, a fim de se verificar este efeito no
polímero final. Observou-se na tamização que o tamanho das partículas no polímero
6 foi menor que 25 µm, como melhor explicado mais adiante, não sendo adequado
para EFS, então vimos que o volume ideal era 20 mL.
Com o volume de solvente ajustado para 20 mL e quantidade fixa de 0,25
mmol de AMI, deu-se início a bateria de síntese dos polímeros numerados de 7 a 13
para avaliar outras modificações, e observar como elas afetariam o rendimento e as
características dos polímeros. Em geral, a proporção de reagentes descrita na
literatura para MIPs sintetizados em EDMA foi de (1:4:20), de acordo com Tamayo et
al. (2007), sendo a proporção usada para MIP/NIP 7. Os polímeros 8 e 9 variaram
somente na proporção de MAA, enquanto que o polímero 10 variou na proporção de
EDMA, comparando com 7. Em relação aos polímeros sintetizados com TRIM 11, 12
e 13, foi usada uma quantidade menor deste em relação ao EDMA, devido a sua
maior capacidade de reticulação. Os polímeros foram sintetizados tendo como base
a proporção usada para o polímero 2 (1:4:8), aumentando um pouco a quantidade
de TRIM chegando a proporção de (1:4:10) para o polímero 11. A partir daí, variou-
se a quantidade de MAA em 12 (1:8:10) e de TRIM em 13 (1:8:5).
Em todas as proporções testadas nos polímeros de 7 a 13 houve
polimerização com bons rendimentos. Os polímeros 9 (1:6:20 EDMA) e 13 (1:8:5
TRIM) apresentaram partículas com aspecto muito heterogêneo, não sendo
adequados para a aplicação como adsorventes em EFS. Os demais polímeros (7, 8,
10, 11 e 12) apresentaram partículas homogêneas para aplicação no cartucho de
EFS, além de bom rendimento. O polímero 10 apresentou maior rigidez,
necessitando de trituração, pois o volume de solvente foi baixo em relação à elevada
quantidade de monômeros (1:8:40), assemelhando-se ao processo de polimerização
em massa, o que não era o proposto para o nosso estudo (precipitação). Mesmo
assim, o polímero foi utilizado para os testes de adsorção seguintes. Os polímeros
sintetizados em EDMA 7 e 8 apresentaram partículas com grumos de aspecto mais
rígido do que os polímeros sintetizados com TRIM, mas não precisaram ser
triturados. As partículas de polímeros sintetizados em TRIM 11 e 12 apresentaram

58
grumos mais soltos e facilmente pulverizáveis, não havendo a necessidade de
trituração e também foram somente tamizados.
A tamização dos polímeros foi necessária para uniformizar as partículas e
avaliar seu tamanho para a aplicação na EFS. O tamanho ideal para EFS situa-se
entre 30 - 60 µm. Partículas menores aumentam a resistência do sistema à
passagem do solvente e exigem a aplicação de muita pressão, como sabemos que
ocorre na cromatografia em fase líquida, onde a fase sólida apresenta partículas de
5 µm. Como a porosidade dos “frits” utilizados nos cartuchos de EFS é de 20 µm, os
polímeros foram peneirados em tamizes de 0,025 a 0,062 mm excluindo-se as
partículas menores ou maiores. O que observou-se visualmente na tamização foi
que as partículas dos polímeros feitos com TRIM apresentaram menor tamanho que
as partículas dos polímeros feitos com EDMA, devido a muitas partículas terem
passado na peneira de 0,032 mm. Isto pode ser devido à menor quantidade deste
agente de reticulação (de 12 a 40 mmol), em comparação ao TRIM (de 4 a 10 mmol)
nos polímeros sintetizados.
Em MIP/NIP 6, onde alterou-se somente o volume de solvente em relação a
MIP/NIP 2 (de 20 para 30 mL), as partículas apresentaram tamanho menor que 25
µm, sendo este inadequado para a aplicação do polímero em EFS. O que pode
explicar este efeito é que um maior volume de solvente reduziu o tamanho da
partícula de polímero formada, pois há maior dificuldade de encontro dos
monômeros com a cadeia polimérica em crescimento. O tamanho de partícula
encontrado na literatura (0,3 a 10 µm) para a técnica de polimerização por
precipitação também utilizada em nosso estudo, pode ser devida a este efeito do
volume do solvente (PÉREZ-MORAL; MAYES, 2004). Manipulando então a
quantidade de solvente, obtivemos partículas maiores para a aplicação dos
polímeros em EFS, sem a necessidade de trituração do polímero final.
A técnica mais utilizada na literatura para síntese de MIPs consiste na
polimerização em massa, principalmente quando aplicada à EFS. No entanto,
existem abordagens que utilizam a técnica por precipitação para aplicação dos
polímeros em EFS (KAREUHANON et al., 2009; ZHANG et al., 2006; CACHO et al.,
2003). A técnica por precipitação foi usada neste trabalho visando a obtenção de
partículas de tamanho mais uniforme, com maior rendimento de sítios de
reconhecimento e assim polímeros com maior capacidade adsortiva, pois a etapa de
trituração é abolida. Mohajeri e colaboradores (2011) observaram que a

59
polimerização por precipitação possibilitou maior reconhecimento e adsorção de
clozapina do que a técnica de polimerização em massa.
O EDMA é o agente de reticulação mais utilizado para a síntese de MIPs,
contudo o uso de TRIM também tem sido investigado, e resultados semelhantes ou
até melhores tem sido obtidos comparados a MIPs sintetizados com EDMA. O TRIM
pode conferir ao MIP formado maior capacidade de adsorção e maior seletividade ao
polímero formado (TARLEY; SOTOMAYOR; KUBOTA, 2005a). Neste trabalho, foi
feita a síntese de polímeros tanto com EDMA quanto com TRIM para melhor
avaliarmos as características do polímero final quanto à adsorção e aplicabilidade
para EFS.
O MAA foi escolhido para ser o monômero funcional para a síntese dos MIPs
de cloridrato de amilorida (AMI), porque possui grupos aceptores (a carbonila do
grupo carboxila, COOH) e doadores (hidroxila) de ligação de hidrogênio capazes de
interagir com a molécula molde, que possui também grupos aceptores (carbonila do
grupo acilguanidina) e doadores (grupos amino) de ligação de hidrogênio (TARLEY;
SOTOMAYOR; KUBOTA, 2005a). Além disso, possui a capacidade de fazer
ligações de natureza iônica devido à presença de grupos ionizáveis (COOH), pois é
um ácido fraco (pKa = 4,7), possibilitando a interação com a AMI que se apresenta
na forma de cátion. A Figura 12 ilustra a interação iônica entre AMI e MAA em uma
síntese de MIP com EDMA ou TRIM como agente de reticulação.
A mistura de solventes de elevada polaridade THF:MeOH:H2O (5:4:1), permite
a síntese dos MIPs para moléculas polares, como descrito por Kareuhanon et al.
(2009). Alguns autores demonstraram o uso de outros solventes polares, como a
acetonitrila, na síntese de MIP por precipitação, a fim de conferir ao polímero maior
capacidade de reconhecimento pela molécula molde, quando aplicados para análise
em sistemas hidrofílicos (YE; CORMACK; MOSBACH, 1999; WANG et al., 2003).
Como o cloridrato de amilorida não foi solúvel em acetonitrila e na mistura
THF:MeOH:H2O (5:4:1) apresentou boa solubilidade, esta foi empregada no
presente estudo.

60
Figura 12. Esquema da síntese de MIP de AMI com EDMA e MAA
Adaptado de BENOS, 1982 e BALAMURUGAN, K.;
PRAKASAM, T.; GOKULAKRISHNAN, 2012
5.2 CARACTERIZAÇÃO DOS POLÍMEROS OBTIDOS
5.2.1 Caracterização química por Espectroscopia no Infravermelho com
Transformada de Fourier (FTIR)
Os espectros de todos os monômeros utilizados na síntese (MAA, TRIM e
EDMA) constam nos Anexos 8.1. Os monômeros ou agentes de reticulação EDMA
e TRIM possuem os mesmos grupamentos químicos e assim, as mesmas bandas
características de monômeros acrílicos, derivados esterificados do ácido acrílico. O
MAA é um ácido também derivado do ácido acrílico. As bandas características de
EDMA e TRIM (ésteres) que podem ser visualizadas com maior evidência estão na
região de 1700 cm-1, banda correspondente ao estiramento de C=O do grupo éster e
a 1150 cm-1, banda correspondente a deformação axial de C-O de éster. Para o
monômero MAA (ácido carboxílico), observa-se uma banda característica de
deformação angular de O-H de ácido carboxílico a 940 cm -1. Também pode ser
observada para o MAA, a banda na região de 1700 cm-1, correspondente ao

61
estiramento de C=O referente a carboxila (COOH), além da banda característica de
deformação axial de C-O de ácido carboxílico de 1200 a 1300 cm-1. Ainda em MAA,
pode-se observar a presença de uma banda larga na região de 2900 cm-1,
característica de deformação axial de O-H de ácido carboxílico, não muito intensa
devido a formação de ligações de hidrogênio intermoleculares. Para o EDMA e
TRIM, nesta mesma região, podem ser vistas bandas de deformação axial de C-H.
Pode-se também observar um sinal em 1670 – 1640 cm-1 proveniente do
estiramento de grupos vinila C=C, presentes em todos os monômeros.
Os espectros das amostras de polímeros MIP/NIP 7, 8, 10, 11 e 12
sintetizados constam no Anexos 8.2.
Nas Figuras 13 (MIP/NIP 7) e 14 (MIP/NIP 11) podemos comparar os
espectros de FTIR de MIP e NIP com os dois agentes de reticulação utilizados no
estudo, sendo o 7 com EDMA e o 11 com o TRIM. Os espectros de FTIR dos
polímeros MIP e NIP em TRIM e EDMA são praticamente idênticos, pois possuem
os mesmos grupamentos químicos. Como a metodologia de síntese de MIP e NIP é
a mesma, os polímeros finais obtidos são os mesmos, exceto o MIP que é
sintetizado na presença de AMI, sendo esta eliminada pela lavagem do polímero.
Assim, a semelhança dos espectros de NIP e MIP demonstra que o fármaco foi
realmente eliminado na fase de lavagem de MIP.
600110016002100260031003600
95,0
95,5
96,0
96,5
97,0
97,5
98,0
98,5
mip7
nip7
Comprimento de onda (cm-1)
Tra
nsm
itânc
ia (
T%
)
Figura 13. Espectros de FTIR de MIP e NIP 7 (EDMA)

62
600110016002100260031003600
88
90
92
94
96
98
MIP 11
NIP 11
Comprimento de onda (cm-1)
Tra
nsm
itânc
ia (
%)
Figura 14. Espectros de FTIR de MIP e NIP 11 (TRIM)
As bandas características dos monômeros utilizados na síntese e que podem
ser visualizadas nos polímeros MIP e NIP são as de EDMA, TRIM e MAA na região
de 1700 cm-1, correspondente ao estiramento de C=O do grupo éster e carboxila do
ácido, de 1150 cm-1 correspondente a deformação axial de C-O de éster e de 1200 a
1300 cm-1 característica de deformação axial de C-O de ácido carboxílico.
Pode-se também observar a ausência do sinal em 1670 – 1640 cm-1
proveniente do estiramento de grupos vinila dos monômeros, que desaparecem nos
polímeros devido a quebra das ligações C=C no processo de polimerização, dando
origem à ligações simples.
Entretanto, não puderam ser observadas com muita evidência bandas na
região de 2900 cm-1 características de estiramento de O-H de ácido carboxílico e C-
H em nenhum dos polímeros sintetizados, predominando assim bandas
características do grupo éster presentes em EDMA e TRIM. Isto pode ser devido a
formação de ligações de hidrogênio intermoleculares que ocorrem entre as cadeias
de polímeros, diminuindo a intensidade das bandas de estiramento de O-H. Além
disso, os monômeros de reticulação EDMA e TRIM estão em excesso em relação ao
monômero funcional MAA, predominando assim as bandas de características de
ésteres.

63
A formação de polímeros porosos altamente reticulados não é uma ciência
inteiramente preditiva e os materiais resultantes são difíceis de caracterizar
completamente, mesmo por ressonância magnética nuclear no estado sólido (RMN)
e espectroscopia no infravermelho por transformada de Fourier (FTIR)
(WHITCOMBE; VULFSON, 2001), assim como através de técnicas de análise
térmica como calorimetria diferencial de varredura (ESFANDYARI-MANESH et al.,
2011).
Não foram observadas bandas características de AMI nos polímeros
sintetizados, principalmente na região de 3250 e 3150 cm-1, como pode ser visto na
Figura 15, correspondente ao estiramento de N-H presente no analito (MAZZO,
1986). Isto confirma a eficiência na lavagem de MIP pela extração no Soxhlet,
eliminando AMI do polímero final.
600110016002100260031003600
80
85
90
95
100
Comprimento de onda (cm-1)
Tra
nsm
itânc
ia (
%)
Figura 15. Espectro de FTIR de AMI
5.2.2 Microscopia Eletrônica de Transmissão de MIP/NIP 12
As imagens obtidas por MET mostram que os polímeros se apresentam na
forma de aglomerados de partículas (Figura 16), similar ao encontrado por alguns
autores (KAREUHANON, et al., 2009; ZHANG et al., 2006). Os polímeros MIP e NIP
possuem morfologia e características de superfície semelhantes sendo muito difícil

64
diferenciá-los, mesmo através por técnicas de porosimetria, que avaliam o tamanho
dos poros formados no polímero, e de determinação da área de superfície (YE;
CORMACK; MOSBACH, 1999).
Figura 16. Imagens de MET dos polímeros NIP (esq.) e MIP (dir.)
Na teoria, a presença da molécula molde, seja qualquer composto, poderia
promover o aumento do tamanho dos poros e da área de superfície dos polímeros,
promovendo diferenças em relação ao polímero não impresso. Diversos trabalhos
descritos na literatura (KAREUHANON, et al., 2009; ZHANG et al., 2006; YE;
CORMACK; MOSBACH, 1999) avaliam estes parâmetros tanto em MIP como em
NIP, mas os autores não chegam a um consenso sobre qual o efeito do analito na
morfologia do polímero impresso em relação ao não impresso.
5.3 ENSAIOS DE ADSORÇÃO
5.3.1 Ensaio de adsorção de cloridrato de amilorida em solução tampão pH 6,5
Os espectros de varredura de cloridrato de amilorida podem ser vistos na
Figura 17. Eles foram obtidos no espectrofotômetro UV-Vis em tampão citrato-
acetato pH 6,5 e HCl 0,1N na faixa de comprimento de onda entre 260 e 390 nm,
utilizando solução de AMI a 9 µg mL-1.

65
260 270 280 290 300 310 320 330 340 350 360 370 380 390 4000
0,1
0,2
0,3
0,4
0,5
0,6Tampão acetato/citrato
HCl 0,1 N
Comprimento de onda (nm)
Abs
Figura 17. Espectro de varredura de cloridrato de amilorida
Os λmáx obtidos em tampão citrato-acetato foram em 285 e 363 nm e idênticos
aos do espectro de varredura feito em HCl 0,1N, solvente preconizado pela
Farmacopéia americana USP XXII (1989) para análise de cloridrato de amilorida.
Sendo assim, as leituras das soluções de AMI em tampão citrato-acetato foram
realizadas a 363 nm.
Para a dosagem por espectrofotometria, a curva de calibração do cloridrato
de amilorida em tampão citrato-acetato pH 6,5 (Figura 18) foi construída na faixa de
concentração de de 0,1 a 20 µg mL-1.
0 2 4 6 8 10 12 14 16 18 20 220
0,2
0,4
0,6
0,8
1
1,2
1,4
f(x) = 0,063x + 0,018R² = 0,995
Concentração (µg/mL)
Ab
s
Figura 18. Curva de calibração AMI em tampão citrato-acetato pH 6,5

66
Os resultados das absorbâncias obtidas de acordo com as concentrações
para a plotagem da curva estão tabelados no Anexo 8.3, item 8.3.1. A curva
apresentou excelente linearidade (R=0,995), como pode ser observado pelo cálculo
da equação da reta presente no gráfico. Os cálculos para o ensaio de adsorção em
tampão foram feitos determinando a concentração livre de AMI no sobrenadante de
acordo com a curva de calibração.
Para determinar a quantidade ideal de polímero a ser utilizada nos ensaios de
adsorção, foram feitos testes variando a quantidade de polímero, na concentração a
9 µg mL-1, em duplicata. Os polímeros testados nesta primeira fase foram MIPs/NIPs
10, 11 e 12, devido a serem os polímeros obtidos com maior rendimento. A
concentração de AMI ligada (adsorvida) no polímero foi calculada subtraindo-se a
concentração inicial pela concentração final de AMI. A partir da concentração livre,
calculou-se a massa de AMI em µg e plotou-se o gráfico de massa de AMI em µg
por mg de polímero. Os resultados das absorbâncias obtidas para os polímeros e os
cálculos da massa de AMI adsorvida em µg constam no Anexo 8.3, item 8.3.2.
Como pode ser observado na Figura 19, a massa de polímero não influenciou
significativamente na massa de AMI adsorvida, por isso se adotou a massa de 7,5
mg para os ensaios de adsorção. Os polímeros MIP/NIP 12 adsorveram maior
massa de AMI do que 10 e 11, além de apresentar resultados mais homogêneos na
adsorção.
7,5 15 300
5
10
15
NIP 10
MIP 10
Massa de po límero (mg)
AM
I a
dso
rvid
a (
µg
)
7,5 15 300
5
10
15
NIP 11
MIP 11
Massa de po límero (mg)
AM
I a
dso
rvid
a (
µg
)
7,5 15 300
5
10
15
NIP 12
MIP 12
Massa de po límero (mg)
AM
I a
dso
rvid
a (
µg
)
Figura 19. Gráficos de adsorção de acordo com a massa de polímero

67
Os polímeros selecionados para os ensaios de adsorção foram os polímeros
MIPs/NIPs 7, 8, 10, 11 e 12, devido a estes apresentarem características mais
satisfatórias para o emprego como adsorventes em EFS.
Definida a quantidade de polímero (7,5 mg), a avaliação da adsorção foi feita
em triplicata na faixa de concentração de AMI de 3 a 15 µg mL -1, segundo a curva de
calibração feita para cloridrato de amilorida em tampão citrato-acetato. A partir da
concentração livre calculada pela curva de calibração, calculou-se a massa de AMI
adsorvida (µg) e plotou-se o gráfico da curva de adsorção Concentração de AMI (µg
mL-1) X Q, onde Q é a quantidade de AMI adsorvida em µg por mg de polímero. Os
resultados das absorbâncias obtidas nos ensaios de adsorção para os polímeros
variando a concentração e os cálculos de Q constam no Anexo 8.3, item 8.3.3.
As curvas de adsorção para os polímeros MIPs/NIPs 7, 8, 10, 11 e 12 na
faixa de concentração de 3 a 15 µg mL-1 são apresentadas na Figura 20. Os valores
de Q máximos (Qmax) obtidos para os polímeros, respectivamente, foram para
MIP/NIP 7, 2,20 e 2,16 µg mg-1; MIP/NIP 8, 2,16 e 2,08 µg mg-1; MIP/NIP 10, 2,37 e
1,90 µg mg-1; MIP/NIP 11, 2,39 e 2,48 µg mg-1 e MIP/NIP 12, 2,52 e 2,77 µg mg-1. Os
polímeros que apresentaram maior capacidade adsortiva para MIP foram os
MIP/NIP 10 (1:8:40 EDMA), 11 (1:4:10 TRIM) e 12 (1:8:10 TRIM), sendo o último
com maior valor de Q. Os dados para MIP/NIP 12 a 3 µg mL-1 não foram realizados e
por isso não constam na curva de adsorção para estes polímeros.
Os resultados dos ensaios de adsorção dos polímeros MIP/NIP 7, 8, 10 e 11
foram submetidos a cálculos estatísticos para avaliação da diferença entre as
médias de Q entre MIP e NIP nas concentrações de 6 a 15 µg mL -1, sendo que para
3 µg mL-1, o cálculo não foi feito devido a grande variabilidade de resultados nesta
concentração. Os cálculos para MIP/NIP 12 não foram feitos pois estes se
apresentavam em duplicata. A princípio foi aplicado o teste F para verificar se as
variâncias entre as amostras de MIP e NIP eram diferentes (resultados não
mostrados). Observou-se que as amostras não eram significativamente diferentes,
para a maioria das concentrações. O teste t de Student foi então aplicado para
comparação entre as médias de Q em MIP e NIP para amostras independentes e
com variâncias iguais, no modo bicaudal ao nível de significância de 5% (α).
As tabelas com os resultados do teste t estatístico aplicado e os respectivos
p-value de MIP e NIP para cada concentração constam no Anexo 8.3, item 8.3.4.

68
3 6 9 12 15
0,0
0,5
1,0
1,5
2,0
2,5
3,0
NIP 7
MIP 7
Concentração µg/mL
Q (
µg
/mg
)
3 6 9 12 15
0,0
0,5
1,0
1,5
2,0
2,5
3,0
NIP 8
MIP 8
Concentração µg/mL
Q (
µg
/mg
)
3 6 9 12 15
0,0
0,5
1,0
1,5
2,0
2,5
3,0
NIP 10
MIP 10
Concentração (µg/mL)
Q (
µg
/mg
)
3 6 9 12 15
0,0
0,5
1,0
1,5
2,0
2,5
3,0
NIP 11
MIP 11
Concentração (µg/mL)
Q (
µg
/mg
)
6 9 12 15
0,0
0,5
1,0
1,5
2,0
2,5
3,0
NIP 12 MIP 12
Concentração (µg/mL)
Q (
µg
/mg
)
Figura 20. Curvas de adsorção de MIPs/NIPs 7, 8, 10, 11 e 12
A hipótese nula (H0) foi aceita em todos os casos, ou seja, as médias de MIP
e NIP são iguais, pois os p-value calculados foram menores que α (0,05) e os
valores de t observados (calculados) foram menores que o tabelado (t=2,776).
Conclui-se assim que os polímeros MIP não apresentaram diferenças significativas
de adsorção comparados aos seus NIP correspondentes na faixa de concentração
de 6 a 15 µg mL-1.
O ensaio de adsorção de MIP/NIP 10 e MIP/NIP 12 foi conduzido de forma a
se avaliar o grau de saturação dos polímeros, ou seja, o máximo de AMI que os

69
polímeros seriam capazes de adsorver. Para isso, os valores de Q foram calculados
para uma faixa de concentração mais ampla que a dos polímeros anteriores,
variando de 6 a 75 µg mL-1 para MIP/NIP 10 e de 6 a 60 µg mL-1 para MIP/NIP 12.
Nos gráficos de adsorção apresentados na Figura 21, pode-se observar os valores
para MIP/NIP 10, onde o Qmax obtido foi de 11,35 (NIP) e 11,45 µg mg-1 (MIP) na
concentração de 75 µg mL-1. Para MIP/NIP 12, o Qmax obtido foi de 10,63 e 10,45 µg
mg-1 na concentração de 60 µg mL-1.
Figura 21. Curvas de adsorção de MIP/NIP 10 e 12 de acordo com a concentração
Observou-se nesses polímeros, que Q aumenta proporcionalmente com o
aumento da concentração, não alcançando a saturação, entretanto também não se
observou diferença entre os MIPs e seus respectivos NIPs. Acima das
concentrações testadas, o cloridrato de amilorida não era completamente
solubilizado, inviabilizando o ensaio em concentrações superiores a 75 µg mL-1.
As taxas médias de adsorção (%) foram calculadas pela razão entre a
quantidade adsorvida e a quantidade inicial de AMI. Os valores encontrados estão
na Tabela 5. A ordem crescente de capacidade média de adsorção foi MIP/NIP 10 <
11 < 8 < 7 < 12. Entre os polímeros com maior capacidade de adsorção, as
proporções usadas na síntese de MIP/NIP 7 (1:4:20 EDMA) foram as ideais para o
agente de reticulação EDMA, confirmando a descrita pela literatura. Quanto ao
MIP/NIP 12 (1:8:10 TRIM), o aumento na capacidade adsortiva pode ter sido devida
não somente à proporção de reagentes, maior do que nos dois estudos localizados
durante a revisão bibliográfica, mas também pela proporção maior de partículas na
faixa de 25 – 32 µm, o que aumenta a superfície de contato. Também não foi
0 10 20 30 40 50 60 70 80
0,0
2,0
4,0
6,0
8,0
10,0
12,0
NIP 10
MIP 10
Concentração (µg/mL)
Q (
µg
/mg
)
0 10 20 30 40 50 60 70 80
0,0
2,0
4,0
6,0
8,0
10,0
12,0
NIP 12
MIP 12
Concentração (µg/mL)
Q (
µg
/mg
)

70
observada diferença aparentemente significativa entre o MIP e seu NIP
correspondente com as proporções testadas quanto às taxas médias de adsorção.
Como o polímero 12 apresentou a maior capacidade média de adsorção, ele foi
escolhido para a confecção do cartucho para EFS e testes de recuperação de AMI.
Vale lembrar que os ensaios de adsorção mantém o fármaco em contato com o
polímero por 16 horas. A passagem pelo cartucho de EFS dura, no máximo, 5
minutos.
Tabela 5. Taxas médias de adsorção (%) dos polímeros
Polímeros Taxas médias de adsorção (%)
MIP 7 76,71
NIP 7 69,53
MIP 8 69,40
NIP 8 70,49
MIP 10 62,31
NIP 10 57,89
MIP 11 64,92
NIP 11 66,28
MIP 12 83,85
NIP 12 88,26
A interação iônica que ocorre entre o cloridrato de amilorida (AMI) e o ácido
metacrílico (MAA) no tampão citrato-acetato pH 6,5 está representada na Figura 22.
Os polímeros possuem ancorados em sua estrutura grupamentos ácidos,
provenientes do MAA (pKa 4,7), livres para interação com a AMI, que possui um pKa
de 8,7. Como o ensaio de adsorção foi feito em tampão citrato-acetato pH 6,5, neste
pH o MAA está carregado negativamente (COO-) e a AMI está na forma de cátion
pois o grupo guanidínico se apresenta com carga positiva, o que favorece a
interação iônica com o polímero e possibilita a adsorção.
Os polímeros sintetizados apresentaram uma boa adsorção, variando de 63 a
88%, o que confirma a interação iônica de AMI com os polímeros, contudo esta não
foi seletiva. A interação não específica dos MIPs com AMI foi possível devido aos
grupos de MAA presentes também no polímero não impresso. A interação do analito
muito polar com o ácido metacrílico pode ter sido prejudicada pela competição com
o solvente de elevada polaridade (THF:MeOH:água) no momento da síntese, que
contém solventes polares como água e metanol em sua composição. O AMI

71
possivelmente interagiu através de ligação de hidrogênio com esses solventes,
explicando a falta de seletividade.
Figura 22. Interação iônica de amilorida com o ácido metacrílico no pH 6,5
Pérez-Moral e Mayes (2004) observaram uma mesma situação de ausência
de seletividade entre MIP e NIP na adsorção de propranolol em uma solução
tamponada a pH 4,6 em MIP sintetizado por precipitação em tolueno. Neste estudo,
os autores fizeram a síntese de MIPs para o propranolol utilizando técnicas de
polimerização diferentes como em massa, por precipitação e por suspensão e
compararam a adsorção do analito no solvente de síntese (tolueno) e em tampão pH
4,6. A falta de seletividade não foi observada neste estudo em outras técnicas de
polimerização como em massa e suspensão para o mesmo analito, somente pela
técnica de precipitação. No caso do propranolol sintetizado por precipitação,
conseguiu-se observar uma diferença na seletividade de MIP em relação a NIP de
50%, no teste de adsorção do analito em tolueno. Como o MIP foi sintetizado em
tolueno, o ambiente se mostrou favorável à interação analito-polímero, assim como
ocorreu na síntese.
Outros autores como Baggiani et al. (2001) e Masqué et al. (2000) utilizaram
solventes polares como acetonitrila e metanol:água 3:1 para a síntese de MIPs
seletivos por polimerização em massa, para análise de herbicidas à base de
derivados de ácidos clorofenoxiacéticos e de p-nitrofenol, respectivamente. Os
autores verificaram a seletividade entre MIP e o polímero não impresso (NIP)
através de análise cromatográfica, utilizando os polímeros como recheio da coluna.
Os fatores de retenção obtidos foram maiores para MIP do que para NIP para os
analitos testados, comprovando a seletividade do polímero e o efeito de impressão

72
molecular. Na polimerização em massa, técnicas usadas por estes autores, o
volume de solvente é muito menor. Isto pode diminuir a interferência que o solvente
empregado na síntese pode ter sobre o analito de interesse, pois os monômeros
estão em excesso em relação ao solvente.
5.3.2 Ensaio de adsorção dos polímeros em THF:MeOH:água (5:4:1)
Para verificarmos se os polímeros sintetizados teriam adsorção no solvente
utilizado na síntese, THF:MeOH:água (5:4:1), foi feito um estudo de adsorção neste
solvente. Os resultados do ensaio de adsorção dos polímeros em THF:MeOH:água
(5:4:1) não foram de acordo com o esperado e descrito na literatura (PÉREZ-
MORAL; MAYES, 2004), pois houve praticamente nenhuma adsorção neste solvente
(resultados não mostrados, absorbâncias semelhantes às soluções controles sem
polímero). O esperado seria uma interação significativa entre MIP e AMI neste
solvente através de ligações de hidrogênio, pois este foi o solvente usado para a
síntese e teoricamente reproduziu-se este ambiente favorável à interação polímero-
AMI. O que poderia explicar este resultado, é que foi utilizado um solvente prótico
que também é capaz de interagir com AMI por ligações de hidrogênio, competindo
com o polímero e dificultando a formação das cavidades seletivas nos MIPs.
Entretanto, a dificuldade na escolha do solvente foi devida a não solubilização de
AMI em solventes orgânicos apróticos, como THF, e inclusive em solventes de
constante dielétrica elevada como a acetonitrila.
Solventes polares apróticos, tais como acetonitrila e THF poderiam ser mais
apropriados para a síntese de MIPs para AMI, mas o sal é praticamente insolúvel
nesses solventes. Spizzirri e Peppas (2005) sintetizaram MIPs para reconhecimento
de colesterol utilizando como solventes porogênicos THF, misturas de THF e DMSO
e THF/água. Neste estudo, os autores observaram que a adição de DMSO
aumentou a capacidade de reconhecimento do polímero impresso para o colesterol,
provavelmente devido a este ser um solvente polar e aprótico, não competindo com
o polímero pela formação das ligações deste com o analito. Assim, podemos sugerir
para uma futura continuação de nosso estudo, a utilização da mistura de solventes
composta por dimetilsulfóxido (DMSO) e THF, ambos solventes apróticos e polares,
sendo uma alternativa para a síntese de MIP para AMI, pois o analito é solúvel em
DMSO.

73
A temperatura da reação (60ºC) também pode influenciar na interação
analito-monômero se esta ocorrer por ligação de hidrogênio. Esta temperatura foi
necessária para a quebra da ligação O-O e consequente liberação dos radicais livres
do peróxido de benzoíla, iniciador usado na síntese dos polímeros, para que este
pudesse dar início à polimerização. Recomenda-se efetuar a polimerização em
baixas temperaturas e, nestas circunstâncias, os iniciadores radicalares ativos
fotoquimicamente por ação de energia UV são os mais indicados, como AIBN.
(TARLEY; SOTOMAYOR; KUBOTA, 2005a).
5.4 EXTRAÇÃO EM FASE SÓLIDA COM MIP (MISPE)
Os cartuchos de EFS foram preenchidos utilizando os polímeros MIP/NIP 12
como adsorvente conforme item 4.3.12, pois a princípio, estes apresentaram maior
capacidade adsortiva nos ensaios de adsorção realizados anteriormente. Os
resultados obtidos nos ensaios de MISPE por espectrofotometria no UV estão
tabelados e constam no Anexo 8.4. Para esses ensaios foi utilizada solução de AMI
a 10 µg mL-1, preparada em tampão citrato-acetato pH 6,5. Uma alíquota foi
colocada (carregada) no cartucho condicionado e, após aplicado o vácuo, o resíduo
em tampão foi recolhido e feita a leitura da absorbância, conforme o item 4.3.13. A
capacidade de retenção foi avaliada pela dosagem de AMI na solução antes
(controle) e após sua passagem pelo cartucho previamente condicionado. Como no
resíduo de carregamento em tampão a absorbância foi próxima de zero, pudemos
concluir que os polímeros foram capazes de reter quase que totalmente o analito.
Na fase de remoção do fármaco adsorvido no cartucho (ou eluição) vários
solventes foram testados, conforme listado na Tabela 6, onde constam os totais de
recuperação calculados (%). Da mesma forma, o solvente foi eluído no cartucho
carregado com AMI sob vácuo e os resíduos recolhidos, evaporados sob N2 e
diluídos em tampão citrato-acetato pH 6,5 para leitura no UV a 363 nm.
A eluição com metanol puro não foi capaz de retirar quantidades significativas
de AMI dos polímeros, obtendo-se uma recuperação de 5,6% para MIP e 9,5% para
NIP. A interação iônica entre o analito e o polímero possui maior força e prevaleceu
em relação à interação entre AMI e metanol por ligação de hidrogênio. Este solvente
poderia ser então usado em uma etapa de lavagem na técnica de MISPE, após o

74
carregamento com tampão, para eliminação de interferentes em uma amostra
contendo AMI, sem que este seja eluído do cartucho.
A eluição total de AMI somente ocorre após lavagem com o solvente na
presença de ácido, que protona o ânion carboxilato de MAA do polímero, rompendo
assim a interação iônica com o analito. Na eluição de AMI com a mistura
metanol:ácido trifluoroacético (TFA) a 0,1%, obteve-se uma recuperação superior a
80% e em TFA a 1% e 2%, a eluição foi total, assim como em metanol:ácido acético
a 1%.
Tabela 6. Recuperação de AMI (%) obtida pela técnica de MISPE
Solvente MIP NIP
Tampão citrato-acetato pH 6,5 0 0
Metanol 5,6 9,5
TFA 0,1% em metanol 89,6 83,3
TFA 1% em metanol >100 >100
TFA 2% em metanol >100 >100
Ácido acético 1% em metanol >100 >100
Para avaliarmos qual a influência de um solvente aprótico na técnica de
MISPE, a acetonitrila foi o solvente escolhido como solvente de carregamento e
eluição. O cartucho, após condicionado conforme o item 4.3.14, foi carregado com
solução preparada em acetonitrila (ACN). Como o AMI não é solúvel diretamente em
ACN, uma solução a 20 µg mL-1 foi preparada diluindo-se uma alíquota da solução
estoque a 1 mg/mL em água ultra-pura com ACN. Após a passagem pelo cartucho,
a solução de acetonitrila foi recolhida, o solvente foi evaporado a 50ºC sob fluxo de
nitrogênio e os resíduos diluídos em tampão citrato-acetato pH 6,5 e quantificados
através das leituras das absorbâncias.
A concentração de AMI que não foi retida correspondeu a cerca de 55% no
NIP e 61% no MIP. Para remover a amilorida do cartucho foi empregada a solução
de ácido acético 1% em ACN, com remoção total do conteúdo adsorvido. Neste
experimento pudemos evidenciar que a interação iônica, ainda que menos
favorecida pela ausência de controle de pH promovido pelo tampão a pH 6,5, foi

75
ainda responsável pela adsorção do fármaco no polímero. Isto ocorreu devido ao
analito estar na forma de cátion mesmo na presença de ACN, devido ao seu elevado
pKa (8,7). O MAA (pKa = 4,7) neste caso está parcialmente carregado, devido ao
condicionamento feito anteriormente em água e por ser um ácido fraco, o que fez
com que a retenção no polímero diminuísse, mas ainda ocorresse a interação de
natureza eletrostática.

6 CONCLUSÕES
Polímeros de impressão molecular foram sintetizados para funcionarem como
adsorventes e aplicados à técnica de extração em fase sólida com impressão
molecular (MISPE), através da confecção de um cartucho contendo os polímeros.
Os MIPs sintetizados foram avaliados quanto à capacidade de adsorção
comparando-se com polímeros sintetizados na ausência da molécula molde (NIP,
polímeros não-impressos). A molécula molde escolhida para a síntese dos MIPs foi
o cloridrato de amilorida (AMI). O polímero com maior capacidade adsortiva,
MIP/NIP 12 foi utilizado na confecção do cartucho e aplicado à extração em fase
sólida como adsorvente, onde observou-se a recuperação de AMI após a eluição no
cartucho.
Vários fatores estão envolvidos na síntese de polímeros de impressão
molecular. A técnica de polimerização, o solvente empregado na síntese e as
proporções dos reagentes de síntese podem modificar as características do
polímero formado e sua capacidade adsortiva. A escolha dos monômeros funcionais
e do agente de reticulação também deve ser avaliada cuidadosamente para cada
analito. Estes fatores devem ser bem estudados para cada molécula molde,
avaliando-se quais são as condições ideais de síntese, considerando também a
aplicação a que se destina o MIP sintetizado.
A polimerização por precipitação é uma técnica de polimerização simples e de
baixo custo que pode, com o adequado controle das condições operacionais,
fornecer partículas de tamanho apropriado para a aplicação na extração em fase
sólida. Esta técnica possibilitou a obtenção de partículas de tamanho adequado para
a extração de AMI por EFS neste estudo.
Na técnica de impressão molecular utilizada neste estudo, as interações
analito-polímero são de natureza não covalente (iônica e por ligação de hidrogênio),

77
e a elevada polaridade de moléculas como o cloridrato de amilorida torna difícil a
escolha de um solvente ideal para a síntese de MIP, sem que este interfira nas
interações com o polímero. Solventes apróticos podem ser mais adequados para a
síntese de MIPs por não possuírem grupos doadores de ligação de hidrogênio,
aumentando as chances de interação entre a molécula molde e o polímero. No
presente estudo, os solventes e condições experimentais testadas não foram
capazes de gerar polímeros com capacidade adsortiva diferenciada entre MIP e NIP,
apesar do polímero ter apresentado um alto grau de adsorção para AMI. Para este
analito, é necessário que a pesquisa continue com a avaliação de outros solventes,
sugerindo em particular a mistura de THF e DMSO.
A técnica de extração em fase sólida com impressão molecular (MISPE) é
uma técnica promissora na área analitica. Ela possibilita a pré-concentração do
analito com o aumento da seletividade, o que pode ser observado amplamente na
literatura, desde que otimizadas as condições de síntese e de EFS para cada
analito.

7 REFERÊNCIAS BIBLIOGRÁFICAS
ABDEL-HAY, Mohamed H. et al. Derivative and derivative ratio spectrophotometric analysis of antihypertensive ternary mixture of amiloride hydrochloride, hydrochlorothiazide and timolol maleate. Journal of Chinese Chemical Society, Taipei: CCS, v. 55, n. 5, p. 971 – 978, out. 2008.
ALLAFCHIAN, Alireza; ENSAFI, Ali Asghar. DNA-based coated wire membrane sensor for selective determination of amiloride in pharmaceutical compounds, plasma and urine. Journal of Brazilian Chemical Society, São Paulo: SBQ, v. 21, n. 3, p. 564 – 570, 2010.
ANDERSSON, Lars I. Molecular imprinting for drug bioanalysis: A review on the application of imprinted polymers to solid-phase extraction and binding assay. Journal of Chromatography B, Amsterdam: Elsevier, v. 739, n. 1, p. 163 – 173, fev. 2000.
AZEVEDO, Sandra M.F.O. et al. Human intoxication by microcystins during renal dialysis treatment in Caruaru – Brazil. Toxicology, Amsterdam: Elsevier, v. 181-182, p. 441-446, dez. 2002.
BAGGIANI, Claudio et al. Molecularly imprinted solid-phase extraction for the clean-up of chlorinated phenoxyacids from aqueous samples. Journal of Chromatography A, Amsterdam: Elsevier, v. 938, n. 1 – 2, p. 35 – 44, dez. 2001.
BALAMURUGAN, Krishnamoorthy; GOKULAKRISHNAN, Kannan; PRAKASAM, Tangirala. Preparation and evaluation of molecularly imprinted polymer liquid chromatography column for the separation of cathine enantiomers. Saudi Pharmaceutical Journal, Amsterdam: Elsevier, v. 20, n. 1, p. 53 – 61, jan. 2012.
BENOS, Dale J. Amiloride: A molecular probe of sodium transport in tissues and cells. American Journal of Physiology, Bethesda: APS, v. 242, n. 3, p. C131 – C145, mar. 1982.
BI, Hongangg; COOPER, Sam F.; CÔTÉ, Michael G. Determination and identification of amiloride in human urine by high-performance liquid chromatography and gas chromatography-mass spectrometry. Journal of Chromatography, Amsterdam: Elsevier, v. 582, n. 1 – 2, p. 93 – 101, nov. 1992.

79
BLOMGREN, Anders et al. Extraction of clenbuterol from calf urine using a molecularly imprinted polymer followed by quantitation by high-performance liquid chromatography with UV detection. Journal of Chromatography A, Amsterdam: Elsevier, v. 975, n. 1, p. 157 – 164, out. 2002.
BOCK, Mark G.; SCHLEGEL, Bernard; SMITH, Graham M. Theoretical estimation of pKa values of pyrazinylguanidine derivatives. The Journal of Organic Chemistry, Washington: ACS, v. 46, n. 9, p. 1925 – 1927, abr. 1981.
CACHO, C. et al. Clean-up of triazines in vegetable extracts by molecularly-imprinted solid-phase extraction using a propazine-imprinted polymer. Analytical and Bioanalytical Chemistry, Nova Iorque: Springer, v. 376, n. 4, p. 491 – 496, jun. 2003.
CAI, Wensheng; GUPTA, Ram B. Molecularly-imprinted polymers selective for tetracycline binding. Separation and Purification Technology, Amsterdam: Elsevier, v. 35, n. 3, p. 215 – 221, mar. 2004.
CANEVAROLO Jr., Sebastião Vicente. Ciência dos polímeros: um texto básico para tecnólogos e engenheiros. 2a edição. São Paulo: Artliber Editora, 2006. 280 p.
CHEN, Wen-Yih; CHEN, Chin-Sung; LIN, Fu-Yung. Molecular recognition in imprinted polymers: thermodinamic investigation of analyte binding using microcalorimetry. Journal of Chromatography A, Amsterdam: Elsevier, v. 923, n. 1 – 2, p. 1 – 6, jul. 2001.
CHIANELLA, Iva et al. MIP-based solid phase extraction cartridges combined with MIP-based sensors for the detection of microcystin-LR. Biosensors and Bioelectronics, Amsterdam: Elsevier, v. 18, n. 2 – 3, p. 119 – 127, mar. 2003.
CORMACK, Peter A.G.; ELORZA, Amaia Zurutuza. Molecularly imprinted polymers: synthesis and characterisation. Journal of Chromatography B, Amsterdam: Elsevier, v. 804, n. 1, p. 173 – 182, mai. 2004.
COSTA, José Luiz da. Extração em fase sólida. In: MOREAU, Regina Lúcia de Moraes; SIQUEIRA, Maria Elisa Pereira Bastos de. Ciências Farmacêuticas: Toxicologia Analítica. Rio de Janeiro: Guanabara Koogan, 2008. 334p. cap. 16, p. 156 – 161.
DOMINGUEZ-VIDAL, A.; ORTEGA-BARRALES, P.; MOLINA-DIAZ, A. Fast flow-injection fluorimetric determination of amiloride by using a solid sensing zone. Talanta, Amsterdam: Elsevier, v. 56, n. 6, p. 1005 – 1013, abr. 2002.
ENSAFI, Ali A.; ALLAFCHIAN, Ali R. Novel and selective potentiometric membrane sensor for amiloride determination in pharmaceutical compounds and urine. Journal of Pharmaceutical and Biomedical Analysis, Amsterdam: Elsevier, v. 47, n. 4 – 5, p. 802 – 806, ago. 2008.
ESFANDYARI-MANESH, Mehdi et al. Effect of porogenic solvent on the morphology, recognition and release properties of carbamazepine-molecularly imprinted polymer nanospheres. Journal of Applied Polymer Science, Nova Jersey: Wiley-Blackwell, v. 121, n. 2, p. 1118 – 1126, jul. 2011.

80
FIGUEIREDO, Eduardo Costa; DIAS, Ana Cristi Basile; ARRUDA, Marco Aurélio Zezzi. Impressão molecular: uma estratégia promissora na elaboração de matrizes para a liberação controlada de fármacos. Revista Brasileira de Ciências Farmacêuticas, São Paulo: USP, v. 44, n. 3, p. 361 – 375, jul. – set. 2008.
FORREST, G. et al. Simple high-performance liquid chromatographic method for the measurement of amiloride in body fluids. Journal of Chromatography, Amsterdam: Elsevier, v. 428, p. 123 – 130, 1988.
GHOLIVAND, Mohammad Bagher; KHODADADIAN, Mehdi; AHMADI, Farhad. Computer aided-molecular design and synthesis of a high selective molecularly imprinted polymer for solid-phase extraction of furosemide from human plasma. Analytica Chimica Acta, Amsterdam: Elsevier, v. 658, n. 2, p. 225 – 232, jan. 2010.
HAGINAKA, Jun et al. Uniform-sized molecularly imprinted polymer for (S)-naproxen selectively modified with hydrophilic external layer. Journal of Chromatography A, Amsterdam: Elsevier, v. 849, n. 2, p. 331 – 339, jul. 1999.
HALVATZIS, Stergios A. et al. Continuous-flow chemiluminometric determination of amiloride and streptomycin by oxidation with IV-bromosuccinimide. Analytica Chimica Acta, Amsterdam: Elsevier, v. 290, n. 1 – 2, p. 172 – 178, mai. 1994.
HUMPAGE, A.R.; MAGALHAES, V.F.; FROSCIO, S.M. Comparison of analytical tools and biological assays for detection of paralytic shellfish poisoning toxins. Analytical and Bioanalytical Chemistry, Nova Iorque: Springer, v. 397, n. 5, p. 1655 – 1671, jul. 2010.
JANGID, Arvind G.; TALE, Rajesh H.; VAIDYA, Vikas V. A single, selective and simple validated method for simultaneous estimation of amiloride and hydrochlorothiazide in human plasma by liquid chromatography – tandem mass spectrometry. Biomedical Chromatography, Nova Jersey: Wiley-Blackwell, v. 26, n. 1, p. 95 – 100, jan. 2012.
JAVANBAKHT, Mehran et al. Solid-phase extraction of tramadol from plasma and urine samples using a novel water-compatible molecularly imprinted polymer. Journal of Chromatography B, Amsterdam: Elsevier, v. 878, n. 20, p. 1700 – 1706, jun. 2010.
JOZWIAKOWSKI, Michael J.; WILLIAMS, Sandy O.; HATHAWAY, Royal D. Relative physical stability of the solid forms of amiloride HCl. International Journal of Pharmaceutics, Amsterdam: Elsevier, v. 91, n. 2 – 3, p. 195 – 207, abr. 1993.
KAREUHANON, Weeranuch et al. Synthesis of molecularly imprinted polymers for nevirapine by dummy template imprinting approach. Cromatographia, Nova Iorque: Springer, v. 70, n. 11 – 12, p. 1531 – 1537, dez. 2009.
KARTAL, Murat; ERK, Nevin. Simultaneous determination of hydrochlorothiazide and amiloride hydrochloride by ratio spectra derivative spectrophotometry and high-performance liquid chromatography. Journal of Pharmaceutical and Biomedical Analysis, Amsterdam: Elsevier, v. 19, n. 3, p. 477 – 485, mar. 1999.

81
LI, Yuan Nian Benny; MOORE, Douglas E.; TATTAM, Bruce N. Photodegradation of amiloride in aqueous solution. International Journal of Pharmaceutics, Amsterdam: Elsevier, v. 183, n.2, p. 109 – 116, jun. 1999.
MAHONY, John O. et al. Molecularly imprinted polymers – potential and challenges in analytical chemistry. Analytica Chimica Acta, Amsterdam: Elsevier, v. 534, n. 1, p. 31 – 39, abr. 2005.
MAIER, Norbert M. et al. Molecularly imprinted polymer-assisted sample clean-up of ochratoxin A from red wine: merits and limitations. Journal of Chromatography B, Amsterdam: Elsevier, v. 804, n. 1, p. 103 – 111, mai. 2004.
MARTÍN, M.E. et al. Partial least-squares method in analysis by differential pulse polarography. Simultaneous determination of amiloride and hydrochlorothiazide in pharmaceutical preparations. Analytica Chimica Acta, Amsterdam: Elsevier, v. 381, n. 2 – 3, p. 247 – 256, fev. 1999.
MASQUÉ, Núria et al. Synthesis and evaluation of a molecularly imprinted polymer for selective on-line solid-phase extraction of 4-nitrophenol from environmental water. Analytical Chemistry, Washington: ACS, v. 72, n. 17, p. 4122 – 4126, set. 2000.
MATSUI, Jun et al. Molecularly imprinted synthetic polymer receptor selective for atrazine. Analytical Chemistry, Washington: ACS, v. 67, n. 23, p. 4404 – 4408, dez. 1995.
MAZZO, David J. Amiloride hydrochloride. In: FLOREY, Klaus. Analytical profiles of drug substances. Orlando: Academic Press, 1986. 796 p., v. 15, p. 1 – 34.
MIRMOMTAZ, E.; ENSAFI, Ali A.; SOLEIMANIAN-ZAD, S. Determination of amiloride using a ds-DNA-modified pencil graphite electrode based on guanine and adenine signals. Electrochimica Acta, Amsterdam: Elsevier, v. 54, n. 3, p. 1141 – 1146, jan. 2009.
MOHAJERI, Seyed Ahmad et al. Clozapine recognition via molecularly imprinted polymers; bulk polymerization versus precipitation method. Journal of Applied Polymer Science, Nova Jersey: Wiley-Blackwell, v. 121, n. 6, p. 3590 – 3595, set. 2011.
PERALTA, Cecilia M.; FERNÁNDEZ, Liliana P.; MASI, Adriana N. A novel application of immobilization on membranes for the separation and spectrofluorimetric quantification of amiloride and furosemide in pharmaceutical samples. Analytica Chimica Acta, Amsterdam: Elsevier, v. 661, n. 1, p. 85 – 90, fev. 2010.
PERALTA, Cecilia M.; FERNÁNDEZ, Liliana P.; MASI, Adriana N. Solid phase extraction using nylon membranes with fluorescence detection as a fast and sensitive method for amiloride and furosemide determination in urine samples. Microchemical Journal, Amsterdam: Elsevier, v. 98, n. 1, p. 39 – 43, mai. 2011.
PEREIRA, Leandro Alves. Desenvolvimento de um método de extração em fase sólida molecularmente impressa (MISPE) para a determinação de fenitrotiona em

82
tomate. Campinas, 2008. 116 f. Dissertação (Mestrado em Química) – Instituto de Química, Universidade Estadual de Campinas, Campinas, 2008.
PÉREZ-MORAL, N.; MAYES, A.G. Comparative study of imprinted polymer particles prepared by different polymerisation methods. Analytica Chimica Acta, Amsterdam: Elsevier, v. 504, n. 1, p. 15 – 21, fev. 2004.
PULGARÍN, José A. Murillo; MOLINA, Aurelia Alañón; LOPEZ, Pablo Fernández. Simultaneous determination of atenolol, propranolol, dipyridamole and amiloride by means of non-linear variable-angle synchronous fluorescence spectrometry. Analytica Chimica Acta, Amsterdam: Elsevier, v. 370, n. 1, p. 9 – 18, ago. 1998.
SCORRANO, Sonia; LONGO, Luigia; VASAPOLLO, Giuseppe. Molecularly imprinted polymers for solid-phase extraction of 1-methyladenosine from human urine. Analytica Chimica Acta, Amsterdam: Elsevier, v. 659, n. 1 – 2, p. 167 – 171, fev. 2010.
SOUZA, Margarita Domingues; BARBOSA, Carlos Maurício. Polímeros com capacidade de reconhecimento molecular no controlo da libertação de fármacos. Parte 1: Síntese e caracterização. Química Nova, São Paulo: SBQ, v. 32, n. 6, p. 1609 – 1619, 2009.
SPIZZIRRI, U. Gianfranco; PEPPAS, Nicholas A. Structural analysis and diffusional behavior of molecularly imprinted polymer networks for cholesterol recognition. Chemistry of Materials, Washington: ACS, v. 17, n. 26, p. 6719 – 6727, dez. 2005.
TAMAYO, F.G., TURIEL, E., MARTÍN-ESTEBAN, A. Molecularly imprinted polymers for solid-phase extraction and solid-phase microextraction: Recent developments and future trends. Journal of Chromatography A, Amsterdam: Elsevier, v. 1152, n. 1 – 2, p. 32 – 40, jun. 2007.
TANDON, Vishnu K.; MAURYA, Hardesh K. 'On Water': unprecedented nucleophilic substitution and addition reactions with 1,4-quinones in aqueous suspension. Tetrahedron Letters, Amsterdam: Elsevier, v. 50, n. 43, p. 5896 – 5902, out. 2009.
TARLEY, César Ricardo Teixeira; SOTOMAYOR, Maria Del Pilar Taboada; KUBOTA, Lauro Tatsuo. Polímeros biomiméticos em química analítica. Parte 1: Preparo e aplicações de MIP (“molecularly imprinted polymers”) em técnicas de extração e separação. Química Nova, São Paulo: SBQ, v. 28, n. 6, p. 1076 – 1086, 2005a.
TARLEY, César Ricardo Teixeira; SOTOMAYOR, Maria Del Pilar Taboada; KUBOTA, Lauro Tatsuo. Polímeros biomiméticos em química analítica. Parte 2: Aplicações de MIP (“molecularly imprinted polymers”) no desenvolvimento de sensores químicos. Química Nova, São Paulo: SBQ, v. 28, n. 6, p. 1087 – 1101, 2005b.
THAKRAL, Seema; MADAN, A.K. Adduction of amiloride hydrochloride in urea through a modified technique for the dissolution enhancement. Journal of Pharmaceutical Sciences, Nova Jersey: Wiley-Blackwell, v. 97, n. 3, p. 1191 – 1201, mar. 2008.

83
THEODORIDIS, Georgios et al. Preparation of a molecularly imprinted polymer for the solid-phase extraction of scopolamine with hyoscyamine as a dummy template molecule. Journal of Chromatography A, Amsterdam: Elsevier, v. 987, n. 1 – 2, p. 103 – 109, fev. 2003.
THEODORIDIS, Georgios; MANESIOTIS, Panagiotis. Selective solid-phase extraction sorbent for caffeine made by molecular imprinting. Journal of Chromatography A, Amsterdam: Elsevier, v. 948, n. 1 – 2, p. 163 – 169, mar. 2002.
TORAL, M. Inés et al. Simultaneous determination of amiloride and furosemide in pharmaceutical formulations by first digital derivative spectrophotometry. International Journal of Pharmaceutics, Amsterdam: Elsevier, v. 249, n. 1 – 2, p. 117 – 126, dez. 2002.
USP XXII – The United States Pharmacopoeia, USP 22. Ed. Rockville: US Pharmacopeial Convention, 1989.
WANG, Dexian et al. Caffeine molecular imprinted microgel spheres by precipitation polymerization. Korean Journal of Chemical Engineering, Nova Iorque: Springer, v. 20, n. 6, p. 1073 – 1076, nov. 2003.
WHITCOMBE, Michael J. ; VULFSON, Evgeny N. Imprinted polymers. Advanced Materials, Nova Jersey: Wiley-Blackwell, v. 13, n. 7, p. 467 – 478, abr. 2001.
WULF, G.; SARHAN, A. Use of polymers with enzyme analogous structures for the resolution of racemates. Angewandte Chemie, Nova Jersey: Wiley-Blackwell, v. 11, n. 4, p. 341 – 344, abr. 1972.
YAN, Mingdi; RAMSTRÖM, Olof. Molecularly Imprinted Materials-Science and Technology. New York: Marcel Dekker, 2005. 752 p.
YE, Lei; CORMACK, Peter A.G.; MOSBACH, Klaus. Molecularly imprinted monodisperse microspheres for competitive radioassay. Analytical Communications, Londres: RSC, v. 36, n. 2, p. 35 – 38, 1999.
ZECEVIC, M. et al. Statistical optimization of a reversed-phase liquid chromatographic method for the analysis of amiloride and hydrochlorothiazide in tablets. Journal of Pharmaceutical and Biomedical Analysis, Amsterdam: Elsevier, v. 22, n. 1, p. 1 – 6, fev. 2000.
ZHANG, Jiang-hua et al. Selective solid-phase extraction of bisphenol A using molecularly imprinted polymers and its application to biological and environmental samples. Analytical and Bioanalytical Chemistry, Nova Iorque: Springer, v. 385, n. 4, p. 780 – 786, jun. 2006.

8 ANEXOS

8.1 ESPECTROS DE FTIR DOS MONÔMEROS
60011001600210026003100360020
40
60
80
100
120
Ácido metacrílico
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
6001100160021002600310036000
20
40
60
80
100
120
EDMA
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
6001100160021002600310036000
20
40
60
80
100
120
TRIM
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)

8.2 ESPECTROS DE FTIR DOS POLÍMEROS
60011001600210026003100360095
95,5
96
96,5
97
97,5
98
98,5
NIP 7
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
60011001600210026003100360095
95,5
96
96,5
97
97,5
98
MIP 7
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
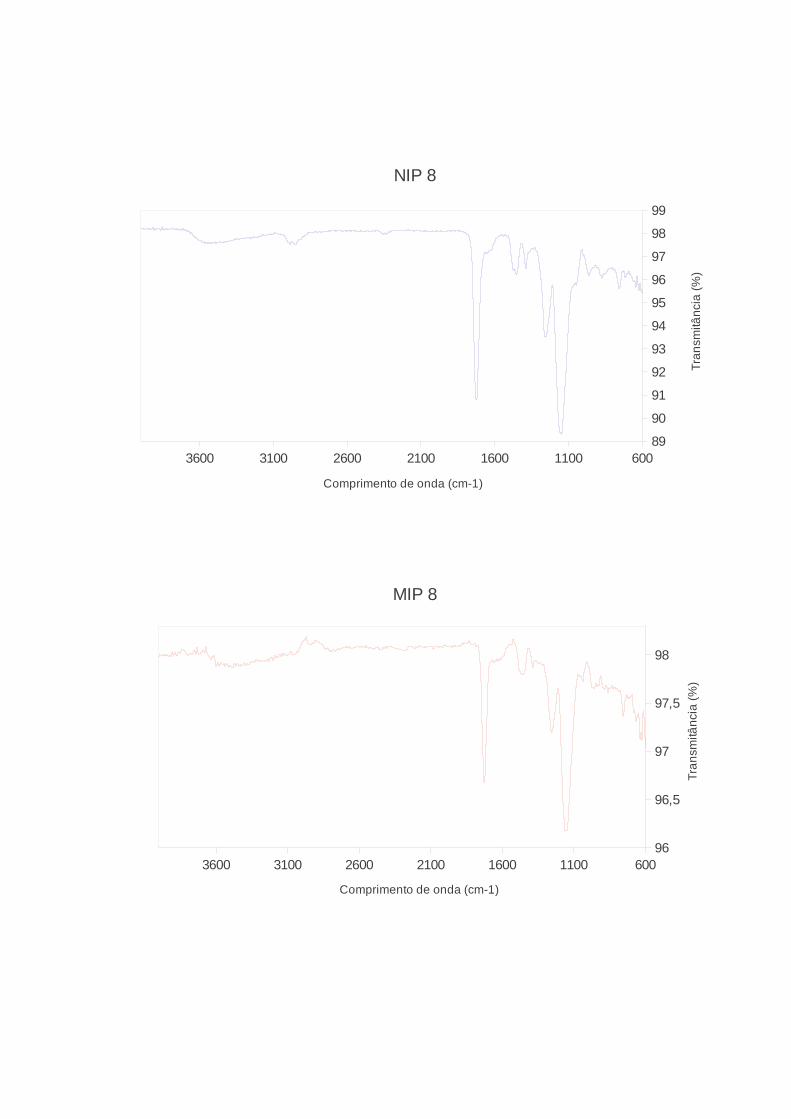
60011001600210026003100360089
90
91
92
93
94
95
96
97
98
99
NIP 8
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
60011001600210026003100360096
96,5
97
97,5
98
MIP 8
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)

60011001600210026003100360086
88
90
92
94
96
98
NIP 10
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
60011001600210026003100360095
95,5
96
96,5
97
97,5
98
98,5
MIP 10
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)

60011001600210026003100360088
90
92
94
96
98
100
NIP 11
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
60011001600210026003100360084
86
88
90
92
94
96
98
100
MIP 11
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)

60011001600210026003100360090
91
92
93
94
95
96
97
98
99
NIP 12
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)
60011001600210026003100360096,6
96,8
97
97,2
97,4
97,6
97,8
98
MIP 12
Comprimento de onda (cm-1)
Tra
nsm
itân
cia
(%
)

8.3 TABELAS COM RESULTADOS DOS ENSAIOS DE ADSORÇÃO
8.3.1 Curva de calibração em tampão citrato-acetato pH 6,5
Abs médias Desvio padrão Coeficiente de variação (%)
0,1 0,012 0,013 0,0032 24,110,1 0,011
0,1 0,0170,3 0,034 0,030 0,0038 12,76
0,3 0,028
0,3 0,0270,5 0,039 0,041 0,0059 14,18
0,5 0,0370,5 0,048
1 0,076 0,075 0,0023 3,09
1 0,0721 0,076
3 0,209 0,209 0,0006 0,283 0,209
3 0,2086 0,398 0,402 0,0047 1,18
6 0,4
6 0,4079 0,606 0,602 0,0047 0,78
9 0,6049 0,597
12 0,799 0,794 0,0044 0,55
12 0,79112 0,792
15 0,992 0,973 0,0186 1,9115 0,955
15 0,97120 1,147 1,220 - -
20 1,292
Concentração (µg mL-1)
8.3.2 Ensaios de adsorção variando a quantidade de polímero
NIP 10
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,266 4,02 7,487,5 9 0,205 3,03 8,9515 9 0,193 2,84 9,24
15 9 0,221 3,29 8,5630 9 0,151 2,16 10,26
30 9 0,108 1,47 11,30
Controle 0,475 7,39P=25,05 mg (bv 25 mL)
AMI adsorvida (µg)
MIP 10
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,238 3,56 8,157,5 9 0,344 5,27 5,59
15 9 0,27 4,08 7,3815 9 0,241 3,61 8,08
30 9 0,279 4,23 7,1630 9 0,31 4,73 6,41
AMI adsorvida (µg)
NIP 11
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,161 2,32 10,02
7,5 9 0,214 3,18 8,73
15 9 0,203 3,00 9,00
15 9 0,145 2,06 10,40
30 9 0,158 2,27 10,09
30 9 0,168 2,44 9,85
Controle 0,510 7,95
P= 10,05 mg (bv 10 mL)
AMI adsorvida (µg)
MIP 11
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,309 4,71 6,447,5 9 0,216 3,21 8,6915 9 0,246 3,69 7,9615 9 0,224 3,34 8,4930 9 0,297 4,52 6,7330 9 0,241 3,61 8,08
AMI adsorvida (µg)

NIP 12
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,127 1,77 10,847,5 9 0,076 0,95 12,07
15 9 0,081 1,03 11,9515 9 0,143 2,03 10,4530 9 0,127 1,77 10,8430 9 0,062 0,73 12,41
AMI adsorvida (µg)
MIP 12
massa (mg) Conc. Inicial Abs Conc. Livre
7,5 9 0,096 1,27 11,597,5 9 0,177 2,58 9,63
15 9 0,093 1,23 11,6615 9 0,092 1,21 11,6930 9 0,105 1,42 11,37
30 9 0,116 1,60 11,10
AMI adsorvida (µg)

8.3.3 Ensaios de adsorção variando a concentração
MIP/NIP 7 (3 a 15 µg/mL)
NIP 7
Abs Q médio Desvio padrão Coeficiente de variação (%)
3 0,095 1,26 2,61 0,35 0,28 - -3 0,137 1,94 1,60 0,213 0,215 3,19 0,01 0,00
6 0,105 1,42 6,87 0,92 0,94 0,03 3,016 0,103 1,39 6,92 0,926 0,089 1,16 7,26 0,979 0,125 1,74 10,89 1,45 1,26 0,19 15,099 0,243 3,65 8,03 1,079 0,184 2,69 9,46 1,2612 0,19 2,79 13,81 1,84 1,92 0,12 6,2312 0,123 1,71 15,44 2,0612 0,184 2,69 13,96 1,8615 0,327 5,00 15,00 2,00 2,16 0,15 6,99
15 0,234 3,50 17,25 2,3015 0,271 4,10 16,35 2,18
Conc. Inicial (µg/mL) Conc. Livre (µg/mL) Amilorida adsorvida (µg) Q* (µg/mg)
*µg de amilorida/mg polímero
MIP 7
Abs Q médio Desvio padrão Coeficiente de variação (%)
3 0,073 0,90 3,15 0,42 0,40 0,02 4,13
3 0,08 1,02 2,98 0,40
3 0,083 1,06 2,90 0,39
6 0,086 1,11 7,33 0,98 0,96 0,08 8,27
6 0,071 0,87 7,69 1,036 0,119 1,65 6,53 0,87
9 0,093 1,23 11,66 1,55 1,49 0,05 3,52
9 0,123 1,71 10,94 1,46
9 0,119 1,65 11,03 1,47
12 0,201 2,97 13,55 1,81 1,92 0,10 5,0112 0,148 2,11 14,83 1,98
12 0,151 2,16 14,76 1,97
15 0,414 6,40 12,90 1,72 2,20 0,42 19,02
15 0,182 2,66 18,51 2,47
15 0,197 2,90 18,15 2,42
Conc. Inicial (µg/mL) Conc. Livre (µg/mL) Amilorida adsorvida (µg) Q* (µg/mg)
*µg de amilorida/mg polímero
NIP 7
Conc. Inicial (µg/mL)* taxa adsorção (%) médias
3,29 2,03 61,70 51,37
3,29 1,35 41,03
3,29 0,10
5,66 4,24 74,91 76,62
5,66 4,27 75,44
5,66 4,50 79,518,85 7,11 80,34 69,60
8,85 5,20 58,87
8,85 6,16 69,60
11,37 8,58 75,46 78,92
11,37 9,66 84,96
11,37 8,68 76,34
14,55 9,55 65,64 71,13
14,55 11,05 75,95
14,55 10,45 71,82
69,53
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)
MIP 7
Conc. Inicial (µg/mL)* taxa adsorção (%) médias
3,29 2,39 72,5463 70,833,29 2,27 69,1146
3,29 2,23 67,6439
5,66 4,55 80,3374 78,635,66 4,79 84,6119
5,66 4,01 70,93358,85 7,62 86,1491 82,75
8,85 7,14 80,68168,85 7,20 81,4106
11,37 8,40 73,8985 78,77
11,37 9,26 81,416911,37 9,21 80,9913
14,55 8,15 55,9916 72,5814,55 11,89 81,7093
14,55 11,65 80,0466
76,71* valores obtidos dos controles
Conc. Adsorvida (µg/mL)

MIP/NIP 8 (3 a 15 µg/mL)
MIP 8
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3 0,113 1,55 2,18 0,29 0,27 - -
3 0,124 1,73 1,91 0,25
3 0,066 0,79 3,31 -
6 0,134 1,89 6,17 0,82 0,83 0,16 18,65
6 0,178 2,60 5,10 0,68
6 0,082 1,05 7,43 0,99
9 0,181 2,65 9,53 1,27 1,27 0,19 14,619 0,239 3,58 8,13 1,08
9 0,124 1,73 10,91 1,45
12 0,256 3,85 12,22 1,63 1,62 0,30 18,58
12 0,355 5,45 9,82 1,31
12 0,169 2,45 14,32 1,91
15 0,205 3,03 17,95 2,39 2,16 0,39 18,03
15 0,417 6,45 12,82 1,7115 0,211 3,13 17,81 2,37
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
NIP 8
Conc. Inicial (µg/mL)* taxa adsorção (%) medias
3,87 1,47 37,98 58,40
3,87 2,43 63,05
3,87 2,87 74,16
6,05 4,49 74,05 74,10
6,05 4,44 73,39
6,05 4,53 74,88
9,35 5,64 60,43 72,83
9,35 7,17 76,79
9,35 7,59 81,28
12,35 9,30 75,38 77,14
12,35 9,06 73,36
12,35 10,20 82,67
15,32 10,43 68,15 69,95
15,32 9,63 62,86
15,32 12,08 78,85
70,49
* valores obtidos dos controles
Conc. Adsorvida (µg/mL) MIP 8Conc. Inicial (µg/mL)* taxa adsorção (%) medias
3,87 2,32 59,99 64,993,87 2,14 55,413,87 3,08 79,586,05 4,16 68,81 69,526,05 3,45 57,086,05 5,00 82,679,35 6,70 71,71 71,659,35 5,77 61,709,35 7,62 81,5412,35 8,50 68,79 68,2612,35 6,90 55,8612,35 9,90 80,1515,32 12,29 80,21 72,5615,32 8,87 57,8915,32 12,19 79,58
69,40* valores obtidos dos controles
Conc. Adsorvida (µg/mL)
NIP 8
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3 0,166 2,40 0,90 0,12 0,22 - -
3 0,106 1,44 2,35 0,31
3 0,079 1,00 0,01 -
6 0,114 1,56 6,65 0,89 0,89 0,01 1,09
6 0,117 1,61 6,58 0,88
6 0,111 1,52 6,73 0,90
9 0,247 3,71 7,94 1,06 1,29 0,21 15,92
9 0,152 2,18 10,23 1,36
9 0,126 1,76 10,86 1,45
12 0,206 3,05 13,43 1,79 1,83 0,12 6,58
12 0,221 3,29 13,06 1,74
12 0,15 2,15 14,78 1,97
15 0,32 4,89 15,17 2,02 2,08 0,25 12,02
15 0,37 5,69 13,96 1,86
15 0,218 3,24 17,64 2,35
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)

MIP/NIP 10 (3 a 15 µg/mL)
NIP 10
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3
33 0,144 2,05 1,43 0,19 0,19
6 0,147 2,10 5,85 0,78 0,86 0,09 11,046 0,09 1,18 7,23 0,96
6 0,131 1,84 6,24 0,839 0,23 3,44 8,35 1,11 1,31 0,17 13,18
9 0,129 1,81 10,79 1,449 0,149 2,13 10,31 1,37
12 0,175 2,55 14,18 1,89 1,74 0,13 7,58
12 0,251 3,77 12,34 1,6512 0,239 3,58 12,63 1,68
15 0,221 3,29 17,56 2,34 1,90 0,43 22,4615 0,486 7,56 11,15 1,49
15 0,363 5,58 14,13 1,88*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
MIP 10
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3 0,191 2,81 0,29 - 0,313
3 0,107 1,45 2,32 0,31
6 0,183 2,68 4,98 0,66 0,76 0,09 12,316 0,15 2,15 5,78 0,77
6 0,125 1,74 6,39 0,85
9 0,27 4,08 7,38 0,98 1,04 0,10 10,079 0,274 4,15 7,28 0,97
9 0,216 3,21 8,69 1,16
12 0,174 2,53 14,20 1,89 1,90 0,03 1,3612 0,165 2,39 14,42 1,92
12 0,181 2,65 14,03 1,87
15 0,216 3,21 17,69 2,36 2,37 0,01 0,59
15 0,209 3,10 17,85 2,3815 0,208 3,08 17,88 2,38
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
NIP 10
Conc. Inicial (µg/mL)* taxa adsorção (%) média
2,58
2,58
2,58 0,53 20,61 20,61
5,56 3,46 62,29 69,35
5,56 4,38 78,82
5,56 3,72 66,93
8,16 4,72 57,90 69,89
8,16 6,35 77,86
8,16 6,03 73,91
10,74 8,19 76,27 69,26
10,74 6,97 64,86
10,74 7,16 66,66
13,82 10,53 76,19 60,36
13,82 6,26 45,26
13,82 8,24 59,62
57,89
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)
MIP 10
Conc. Inicial (µg/mL)* taxa adsorção (%) média
2,582,582,58 1,13 43,74 43,745,56 2,88 51,84 60,645,56 3,41 61,425,56 3,82 68,678,16 4,08 49,99 53,298,16 4,01 49,208,16 4,95 60,6710,74 8,21 76,42 76,5210,74 8,35 77,7710,74 8,09 75,3713,82 10,61 76,78 77,3613,82 10,72 77,5913,82 10,74 77,71 62,31
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)
MIP/NIP 10 (6 a 75 µg/mL)
NIP 10
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio
6 0,206 3,05 4,43 0,59 0,69
6 0,147 2,10 5,85 0,7815 0,248 3,73 16,91 2,25 2,2015 0,284 4,31 16,04 2,14
30 0,415 6,42 35,37 4,72 4,6730 0,445 6,90 34,65 4,62
75 1,16 18,44 84,85 11,31 11,35
75 1,14 18,11 85,33 11,38
Amilorida adsorvida (µg) Q* (µg/mg)
MIP 10
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio
6 0,183 2,68 4,98 0,66 0,776 0,115 1,58 6,63 0,8815 0,231 3,45 17,32 2,31 2,3615 0,198 2,92 18,12 2,4230 0,377 5,81 36,29 4,84 4,9130 0,335 5,13 37,31 4,9775 1,042 16,53 87,70 11,69 11,7175 1,03 16,34 87,99 11,73
Amilorida adsorvida (µg) Q* (µg/mg)

MIP/NIP 11 (3 a 15 µg/mL)
NIP11
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3 0,145 2,06 1,40 0,19 0,23 - -
3 0,221
3 0,117 1,61 2,08 0,28
6 0,145 2,06 5,90 0,79 0,80 0,01 1,63
6 0,142 2,02 5,98 0,80
6 0,137 1,94 6,10 0,81
9 0,24 3,60 8,10 1,08 1,24 0,15 11,82
9 0,179 2,61 9,58 1,28
9 0,151 2,16 10,26 1,37
12 0,175 2,55 14,18 1,89 1,89 0,08 4,18
12 0,198 2,92 13,62 1,82
12 0,149 2,13 14,81 1,97
15 0,189 2,77 18,34 2,45 2,48 0,04 1,56
15 0,178 2,60 18,60 2,48
15 0,165 2,39 18,92 2,52
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
MIP11
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio Desvio padrão Coeficiente de variação (%)
3 0,172 2,50 0,75 0,10 0,22 - -
3 0,196 2,89 - -
3 0,1 1,34 2,49 0,33
6 0,109 1,48 6,77 0,90 0,82 0,11 13,01
6 0,123 1,71 6,44 0,86
6 0,172 2,50 5,25 0,70
9 0,203 3,00 9,00 1,20 1,28 0,07 5,30
9 0,164 2,37 9,94 1,33
9 0,17 2,47 9,80 1,31
12 0,223 3,32 13,02 1,74 1,78 0,07 3,76
12 0,216 3,21 13,19 1,76
12 0,184 2,69 13,96 1,86
15 0,224 3,34 17,49 2,33 2,39 0,06 2,36
15 0,189 2,77 18,34 2,45
15 0,207 3,06 17,90 2,39
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
NIP 11
Conc. Inicial (µg/mL)* taxa adsorção (%) média
2,9 0,84 28,97 36,722,92,9 1,29 44,485,81 3,75 64,54 65,46
5,81 3,79 65,235,81 3,87 66,618,77 5,17 58,95 68,198,77 6,16 70,24
8,77 6,61 75,3711,4 8,85 77,63 77,7811,4 8,48 74,3911,4 9,27 81,3215,44 12,67 82,06 83,25
15,44 12,84 83,1615,44 13,05 84,52
66,28
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)
MIP 11
Conc. Inicial (µg/mL)* taxa adsorção (%) media
2,9 0,4 13,79 33,79
2,9
2,9 1,56 53,79
5,81 4,33 74,53 67,36
5,81 4,1 70,57
5,81 3,31 56,97
8,77 5,77 65,79 70,20
8,77 6,4 72,98
8,77 6,3 71,84
11,4 8,08 70,88 73,04
11,4 8,19 71,84
11,4 8,71 76,40
15,44 12,1 78,37 80,20
15,44 12,67 82,06
15,44 12,38 80,18
64,92
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)

MIP/NIP 12 (6 a 60 µg/mL)
NIP 12
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio
6 0,063 0,74 7,89 1,05 1,066 0,056 0,63 8,06 1,0712 0,074 0,92 16,62 2,22 2,2112 0,077 0,97 16,55 2,2115 0,091 1,19 20,71 2,76 2,7715 0,086 1,11 20,83 2,78
22,5 0,145 2,06 30,65 4,09 3,9822,5 0,209 3,10 29,10 3,8830 0,244 3,66 39,51 5,27 5,3630 0,185 2,71 40,94 5,46
37,5 0,314 4,79 49,06 6,54 6,6237,5 0,268 4,05 50,18 6,6945 0,349 5,35 59,47 7,93 7,9245 0,355 5,45 59,32 7,91
52,5 0,462 7,18 67,98 9,06 9,1352,5 0,423 6,55 68,93 9,1960 0,447 6,94 79,60 10,61 10,6360 0,435 6,74 79,89 10,65
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
MIP 12
Conc. Inicial (µg/mL) Abs Conc. Livre (µg/mL) Q médio
6 0,132 1,85 6,22 0,83 0,85
6 0,116 1,60 6,60 0,88
12 0,14 1,98 15,02 2,00 2,00
12 0,145 2,06 14,90 1,99
15 0,169 2,45 18,82 2,51 2,52
15 0,163 2,35 18,97 2,53
22,5 0,184 2,69 29,71 3,96 3,82
22,5 0,274 4,15 27,53 3,67
30 0,244 3,66 39,51 5,27 5,2730 0,241 3,61 39,58 5,28
37,5 0,293 4,45 49,57 6,61 6,63
37,5 0,278 4,21 49,94 6,66
45 0,374 5,76 58,86 7,85 7,88
45 0,357 5,48 59,27 7,90
52,5 0,418 6,47 69,05 9,21 9,17
52,5 0,442 6,85 68,47 9,13
60 0,491 7,65 78,53 10,47 10,45
60 0,505 7,87 78,19 10,43
*µg de amilorida/mg polímero
Amilorida adsorvida (µg) Q* (µg/mg)
MIP 12
Conc. Inicial (µg/mL)* taxa adsorção (%) médias
5,92 4,06 68,66 68,66
5,92 4,32 73,02
11,63 9,65 82,94 82,94
11,63 9,56 82,25
14,11 11,66 82,63 82,63
14,11 11,76 83,31
21,17 18,48 87,28 87,28
21,17 17,02 80,42
26,77 23,11 86,32 86,32
26,77 23,16 86,50
34,77 30,32 87,20 87,20
34,77 30,56 87,89
42,26 36,50 86,37 86,37
42,26 36,78 87,02
49,1 42,63 86,83 86,83
49,1 42,25 86,04
56,45 48,80 86,46 86,46
56,45 48,58 86,06
83,85
* valores obtidos dos controles
Conc. Adsorvida (µg/mL)NIP 12
Conc. Inicial (µg/mL)* taxa adsorção (%) médias
5,92 5,18 87,47 87,475,92 5,29 89,37
11,63 10,71 92,09 92,0911,63 10,66 91,6814,11 12,92 91,54 91,54
14,11 13,00 92,1121,17 19,11 90,25 90,25
21,17 18,07 85,3726,77 23,11 86,32 86,3226,77 24,06 89,88
34,77 29,98 86,22 86,2234,77 30,72 88,3642,26 36,91 87,33 87,33
42,26 36,81 87,1049,1 41,92 85,38 85,38
49,1 42,55 86,6656,45 49,51 87,71 87,7156,45 49,71 88,06
88,26* valores obtidos dos controles
Conc. Adsorvida (µg/mL)

8.3.4 Análise estatística com os resultados dos ensaios de adsorção (Teste t)
Teste t de Student para amostras independentes com variancias iguais
n1 = n2 = 3graus lib. = 4 H0 = médias iguais
H1 = médias diferentest tab = 2,776
MIP/NIP 7 MIP/NIP 8 MIP/NIP 10 MIP/NIP 11
6 µg/mLmédia MIP 0,96 0,83 0,76 0,82média NIP 0,94 0,89 0,86 0,8dp MIP 0,08 0,16 0,09 0,11dp NIP 0,03 0,01 0,09 0,01t calculado 0,049 0,668 1,361 0,314P-value 0,666 0,57 0,277 0,746Decisão Aceita-se H0 Aceita-se H0 Aceita-se H0 Aceita-se H0
9 µg/mLmédia MIP 1,49 1,27 1,04 1,28média NIP 1,26 1,29 1,31 1,24dp MIP 0,05 0,19 0,1 0,07dp NIP 0,19 0,21 0,17 0,15t calculado 2,027 0,122 2,371 0,418P-value 0,110 0,900 0,081 0,723Decisão Aceita-se H0 Aceita-se H0 Aceita-se H0 Aceita-se H0
12 µg/mLmédia MIP 1,92 1,62 1,9 1,78média NIP 1,92 1,83 1,74 1,89dp MIP 0,1 0,3 0,03 0,07dp NIP 0,12 0,12 0,13 0,08t calculado 0 1,128 2,077 1,79P-value 0,973 0,310 0,115 0,144Decisão Aceita-se H0 Aceita-se H0 Aceita-se H0 Aceita-se H0
15 µg/mLmédia MIP 2,2 2,16 2,37 2,39média NIP 2,16 2,08 1,9 2,48dp MIP 0,42 0,39 0,01 0,06dp NIP 0,15 0,25 0,43 0,04t calculado 0,155 0,297 1,893 2,162P-value 0,878 0,780 0,130 0,075Decisão Aceita-se H0 Aceita-se H0 Aceita-se H0 Aceita-se H0
α = 0,05Decisão: t calc < t tab e p-value > α = Aceita-se H0

8.4 TABELAS COM RESULTADOS DA EXTRAÇÃO EM FASE SÓLIDA COM OS
POLÍMEROS (MISPE)
NIP 12
Fração Abs Massa final (µg) Recuperação (%) médias
19,94 0,635 9,97
Carregamento (2 mL) 0,004 -0,21 0 0 0
tampão citrato-acetato pH 6,5 0 -0,27 0 0 0
0 -0,27 0 0 0
0,006 -0,18 0 0 0Lavagem* (2 mL)
Metanol 0,076 0,95 1,9 9,5 9,5
Eluição* (2 mL)Metanol:TFA 1% 0,686 10,79 21,58 108,2 >100
Metanol:TFA 1% 0,73 11,50 23 115,4 >100
Metanol:TFA 2% 0,779 12,29 24,58 123,3 >100
19,06 0,608 9,53
Carregamento (2 mL) 0 0 0 0 0
tampão citrato-acetato pH 6,5
Eluição* (2 mL)
Metanol:TFA 0,1% 0,451 7,00 14 73,5 83,3Metanol:TFA 0,1% 0,567 8,87 17,74 93,1
Metanol:HAc 1% 0,63 9,89 19,78 103,8 >100
* etapas realizadas em cada replicata do cartucho (separadamente) após o carregamento
Fração Abs Massa final (µg) Recuperação (%) médias
15,27 0,964 15,27
Lavagem* (1 mL)
ACN 0,548 8,56 8,56 56,1 54,8
ACN 0,523 8,16 8,16 53,4
Eluição* (2 mL)
ACN: HAc 1% 0,235 3,52 7,04 46,1 50,4
ACN: HAc 1% 0,276 4,18 8,36 54,8
ACN: HAc 2% 0,008 -0,15 0 0 0
ACN: HAc 2% 0,004 -0,21 0 0 0Total recuperação (%) 105,2
* etapas realizadas na mesma replicata do cartucho após o carregamento
massa inicial (µg) Conc. final (µg/mL)
Amostra (10 µg/mL) 2 mL
Amostra (10 µg/mL) 2 mL
massa inicial (µg) Conc. final (µg/mL)
Amostra (20 µg/mL) 1 mL

MIP 12
Fração Abs Massa final (µg) Recuperação (%) médias
19,94 0,635 9,97
Carregamento (2 mL) 0,003 -0,23 0 0 0
tampão citrato-acetato pH 6,5 0,014 -0,05 0 0 0
0,006 -0,18 0 0 0
0 -0,27 0 0 0
Lavagem (2 mL)
Metanol 0,052 0,56 1,12 5,6 5,6
Eluição (2 mL)
Metanol:TFA 1% 0,714 11,24 22,48 112,7 >100
Metanol:TFA 1% 0,751 11,84 23,68 118,8
Metanol:TFA 2% 0,782 12,34 24,68 123,8 >100
Metanol:TFA 2% 0,756 11,92 23,84 119,6
19,06 0,608 9,53
Carregamento (2 mL) 0 0,00 0 0 0
tampão citrato-acetato pH 6,5
Eluição (2 mL)
Metanol:TFA 0,1% 0,579 9,06 18,12 95,1 89,6
Metanol:TFA 0,1% 0,514 8,02 16,04 84,2
Metanol:HAc 1% 0,656 10,31 20,62 108,2 >100
Metanol:HAc 1% 0,621 9,74 19,48 102,2
* etapas realizadas em cada replicata do cartucho (separadamente) após o carregamento
Fração Abs Massa final (µg) Recuperação (%) médias
15,27 0,964 15,27
Lavagem (1 mL)
ACN 0,422 6,53 6,53 42,8 61,5
ACN 0,778 12,27 12,27 80,4
Eluição (2 mL)
ACN: HAc 1% 0,195 2,87 5,74 37,6 35,6
ACN: HAc 1% 0,176 2,56 5,12 33,5
ACN: HAc 2% 0,013 -0,06 0 0 0
ACN: HAc 2% 0,008 -0,15 0 0
Total recuperação (%) 97,1
* etapas realizadas na mesma replicata do cartucho após o carregamento
massa inicial (µg) Conc. final (µg/mL)
Amostra (10 µg/mL) 2 mL
Amostra (10 µg/mL) 2 mL
massa inicial (µg) Conc. final (µg/mL)
Amostra (20 µg/mL) 1 mL


















