
VERIFICAÇÃO DA ESTABILIDADE ESTRUTURAL DO...
19
1 VERIFICAÇÃO DA ESTABILIDADE ESTRUTURAL DO PEPTÍDEO CN-AMP1 POR MEIO DE DINÂMICA MOLECULAR (VERIFICATION OF THE STRUCTURAL STABILITY OF THE PEPTIDE CN-AMP1 THROUGH MOLECULAR DYNAMICS) André Luiz Raimundo da Cruz 1 , Diego O. Nolasco 1,2 1 Curso de Física – Universidade Católica de Brasília 2 Programa de Pós Graduação em Ciências Genômicas e Biotecnologia – Universidade Católica de Brasília Resumo Dinâmica molecular é uma técnica que pode ajudar a entender vários problemas físicos que, ao longo dos tempos, mostraram-se trabalhosos quando se tem a necessidade de realizar simulações. Criada no final da década de 50 por Alder e Wainwright para estudar as interações entre esferas rígidas, o método de dinâmica molecular se mostrou revolucionário com Rahman e Stillinger em 1964 quando ambos realizaram a primeira simulação utilizando um potencial realístico de argônio liquido. Neste trabalho temos como objetivo verificar a estabilidade estrutural do peptídeo Cn-AMP1 por meio de dinâmica molecular. Palavras-chave: Peptídeo, CN-AMP1, dinâmica molecular, conformação estrutural. ABSTRACT Molecular dynamics is a technique that may help in understanding many physical problems that showed, beyond times, a bit cumbersome when you have the need to do a simulation or even some tests. Built in the late 50’s by Alder and Wainwri ght to study the interactions between hard spheres, the method of molecular dynamics has proved to be revolutionary in Rahman and Stillinger 1964 job when they performed the first simulation using a realistic potential for liquid argon. In this work we aim to verify the structural stability of the Cn- AMP1 peptide using molecular dynamics. keywords: Peptide, CN-AMP1, molecular dynamics, structural conformation. 1. Introdução Por que temos tantos problemas para combater o câncer? Por que alguns antibióticos não funcionam mais com algumas bactérias? São essas e outras perguntas relacionadas ao assunto que movem a ciência para a necessidade de desenvolver métodos alternativos para tais problemas. Neste trabalho serão realizadas duas simulações computacionais, a fim de verificar qual estrutura adotada pelo peptídeo Cn-AMP1, bem como uma analise de gráficos construídos com base nos dados adquiridos nas simulações. Na primeira simulação será utilizado um solvente contendo água e TFE, já na segunda simulação o solvente utilizado contém apenas água. Em ambos os casos o peptídeo Cn-AMP1 será inserido nestes sistemas a fim de interagir eletrostaticamente para que seja possível observar qual a estrutura adotada pelo mesmo ao final da simulação. Administrar uma droga capaz de combater algumas bactérias e até mesmo o câncer, sem todo o sofrimento que os tratamentos atuais trazem ao
Transcript of VERIFICAÇÃO DA ESTABILIDADE ESTRUTURAL DO...

1
VERIFICAÇÃO DA ESTABILIDADE ESTRUTURAL DO
PEPTÍDEO CN-AMP1 POR MEIO DE DINÂMICA MOLECULAR
(VERIFICATION OF THE STRUCTURAL STABILITY OF THE PEPTIDE CN-AMP1 THROUGH
MOLECULAR DYNAMICS)
André Luiz Raimundo da Cruz1, Diego O. Nolasco1,2
1Curso de Física – Universidade Católica de Brasília 2Programa de Pós Graduação em Ciências Genômicas e Biotecnologia – Universidade
Católica de Brasília
Resumo Dinâmica molecular é uma técnica que pode ajudar a entender vários problemas físicos
que, ao longo dos tempos, mostraram-se trabalhosos quando se tem a necessidade de realizar
simulações. Criada no final da década de 50 por Alder e Wainwright para estudar as interações
entre esferas rígidas, o método de dinâmica molecular se mostrou revolucionário com Rahman
e Stillinger em 1964 quando ambos realizaram a primeira simulação utilizando um potencial
realístico de argônio liquido. Neste trabalho temos como objetivo verificar a estabilidade
estrutural do peptídeo Cn-AMP1 por meio de dinâmica molecular.
Palavras-chave: Peptídeo, CN-AMP1, dinâmica molecular, conformação estrutural.
ABSTRACT Molecular dynamics is a technique that may help in understanding many physical
problems that showed, beyond times, a bit cumbersome when you have the need to do a
simulation or even some tests. Built in the late 50’s by Alder and Wainwright to study the
interactions between hard spheres, the method of molecular dynamics has proved to be
revolutionary in Rahman and Stillinger 1964 job when they performed the first simulation using
a realistic potential for liquid argon. In this work we aim to verify the structural stability of the Cn-
AMP1 peptide using molecular dynamics.
keywords: Peptide, CN-AMP1, molecular dynamics, structural conformation.
1. Introdução
Por que temos tantos problemas para combater o câncer? Por que
alguns antibióticos não funcionam mais com algumas bactérias? São essas e
outras perguntas relacionadas ao assunto que movem a ciência para a
necessidade de desenvolver métodos alternativos para tais problemas.
Neste trabalho serão realizadas duas simulações computacionais, a fim
de verificar qual estrutura adotada pelo peptídeo Cn-AMP1, bem como uma
analise de gráficos construídos com base nos dados adquiridos nas
simulações. Na primeira simulação será utilizado um solvente contendo água e
TFE, já na segunda simulação o solvente utilizado contém apenas água.
Em ambos os casos o peptídeo Cn-AMP1 será inserido nestes sistemas
a fim de interagir eletrostaticamente para que seja possível observar qual a
estrutura adotada pelo mesmo ao final da simulação.
Administrar uma droga capaz de combater algumas bactérias e até
mesmo o câncer, sem todo o sofrimento que os tratamentos atuais trazem ao

2
paciente, é um dos maiores objetivos da física médica. Atualmente já é
possível afirmar que o uso de antibióticos no combate as bactérias já está
ultrapassado e tem vida curta, pelo fato destes seres possuírem vários
mecanismos que os tornam capazes de burlar a eficiência dos antibióticos.
Já o câncer é a segunda maior causa de morte (segundo a OMS) e
possui um tratamento doloroso tanto físico quanto psicológico podendo
provocar, diarreia, feridas na boca, queda de cabelo, enjoo dentre outros
desconfortos. A estimativa é que o câncer só neste ano matará mais de meio
milhão de pessoas. No Brasil o câncer é a segunda maior causa de morte, logo
atrás das doenças cardiovasculares de acordo com os levantamentos do
instituto nacional de câncer.
O peptídeo Cn-AMP1 encontrado na água do coco verde, possui a
capacidade de interagir com a membrana celular e, dessa forma, destruir
células cancerosas e bactérias. O peptídeo Cn-AMP1 é um peptídeo catiônico
curto que tem toxidade seletiva para as células cancerosas, mas não possui
nenhuma atividade hemolítica, apresenta também ação antimicrobiana potente
tanto contra bactérias Gram-positivas quanto para bactérias Gram-negativas
(SILVA, 2012).
2. Objetivo
Este trabalho tem como objetivo, verificar por meio de simulação
computacional qual é a estrutura adotada pelo peptídeo Cn_AMP1 em um
solvente contendo água e verificar também esta estrutura em um solvente
contendo água e TFE. Para a realização do trabalho foram realizadas duas
simulações computacionais, uma contendo somente o peptídeo e água e a
segunda simulação contendo o peptídeo água e TFE.
3. Referencial teórico
3.1. Dinâmica molecular clássica
A dinâmica molecular consiste na observação da evolução temporal do
sistema em estudo. A análise das trajetórias fornece suporte para o
entendimento e compreensão dos fatores que contribuem para a variabilidade
e estabilidade conformacional da proteína (NAMBA et al., 2008).

3
A simulação de dinâmica molecular consiste da solução numérica, passo
a passo, da equação de movimento de Newton, que pode ser descrita para um
sistema por.
,
Sendo este um modelo clássico, pois todos os átomos do sistema são
encarados pelo programa como esferas rígidas, com raios obtidos a partir de
medidas ou valores teóricos. Os átomos têm uma carga líquida, obtida da
teoria, sendo que estes átomos interagem eletrostaticamente como "molas"
obedecendo a lei de Robert Hoock. As interações são representadas por
potenciais clássicos.
As forças são a derivada de um potencial elétrico V( , , ..., ), da
forma:
= -
·.
Integrando-se as equações de movimento, obtemos as velocidades, cuja
integral, por sua vez, proporciona a mudança de posição do átomo. Com as
novas posições e velocidades de cada átomo, obtêm-se a energia potencial e
cinética do sistema. Aplicando-se sucessivamente esse procedimento, obtém-
se o que se denomina de “trajetória”, que nada mais é do que o conjunto de
posições e velocidades de cada partícula ao longo do tempo. As equações são
resolvidas simultaneamente em todos os passos da dinâmica, enquanto a
temperatura do sistema é controlada por meio de um banho térmico, a pressão
e volume são mantidos constantes (Namba, Silva, & Silva, 2008).
3.2. Simulação computacional
Na ciência é necessário o desenvolvimento de modelos, modelos estes
que vão organizar um comportamento natural (GAVIRA, 2003). Mas para se
construir um modelo é indispensável à realização de exaustivas observações e
testes. Mas nem sempre observar ou testar é uma tarefa simples, podendo ser
simplificada com a ajuda de recursos computacionais.
Os investimentos em novas tecnologias são altos e arriscados, porém
essenciais para o desenvolvimento científico. O desenvolvimento de recursos
que garantam a eficácia desses investimentos e reduzam riscos é de extrema
importância. Um desses recursos é a simulação computacional, a ciência

4
computacional utiliza o conhecimento sobre determinado problema e o
incorpora em um modelo matemático, que fornece mais informações sobre o
mesmo. A importância dessas simulações se dá por ser um método barato e
fornece informações extras que auxiliam no planejamento e na
interpretação dos experimentos (Gavira, 2003).
3.3. GROMACS
GROMACS é um pacote versátil para realizar dinâmica molecular. O
programa simula as equações do movimento de Newton utilizando o campo de
força GROMACS e condições periódicas de contorno para sistemas com
centenas de milhões de partículas, sendo este um trabalho quase impossível
manualmente. O programa é projetado principalmente para moléculas
bioquímicas como proteínas, lipídios e ácidos nucleicos que possuem várias
interações complexas. O pacote computacional GROMACS é extremamente
rápido para calcular essas interações.
3.4. TFE
O trifluoretanol é um solvente capaz de induzir a proteína a adotar uma
estrutura em alfa hélice. O TFE é um co-solvente anfipático, que possui uma
região polar e outra apolar tendo portanto facilidade para se aglomerar em
solução aquosa, proporcionando um ambiente ao mesmo tempo hidrofóbico e
hidrofílico a proteína (figura 1). O grupo polar OH do TFE pode formar ligações
de hidrogênio com outras moléculas de TFE, com água e outros grupos polares
do peptídeo, enquanto o grupo CF3 protege as regiões hidrofóbicas da proteína
simulando assim o ambiente de uma membrana celular (NOLASCO, 2010).
Figura 1: Trifluoretanol é o composto orgânico representado pela fórmula CF3CH2OH. Esta
molécula apresenta características anfipáticas, pois os átomos de flúor (representados pelas
esferas amarelas) possuem facilidade para se ligar às moléculas hidrofílicas. Já o grupo OH

5
(representado pelas esferas vermelha e brancas) possui facilidade em se ligar às moléculas
hidrofóbicas.
3.5 Proteína
As proteínas são as moléculas orgânicas mais abundantes e importantes
nas células, estando presentes em todas as partes de um organismo, outro fato
importante a cerca das proteínas é a diversidade de funções biológicas que as
mesmas podem desempenhar, com propriedades e atividades fantasticamente
distintas, como em músculos, cabelos, unhas, penas de pássaros, anticorpos e
uma série de outros exemplos, cada qual desempenhando uma função
biológica característica (CURTIS helena, 1997. P.72-73.).
As proteínas pertencem à classe dos peptídeos, pois são formadas por
aminoácidos ligados entre si por ligações peptídicas (CURTIS helena, 1997.
P.72-73.). Uma ligação peptídica é a união do grupo amino (-NH2) de um
aminoácido com o grupo carboxila (-COOH) de outro aminoácido, através da
formação de uma amida.
As proteínas são polímeros cujas unidades fundamentais são os
aminoácidos. Os aminoácidos, por sua vez, são moléculas orgânicas as quais
possuem ligadas ao mesmo átomo de carbono (denominado de carbono α) um
átomo de hidrogênio, um grupo amina, um grupo carboxílico e uma cadeia
lateral “R” característica para cada aminoácido. É o radical "R" quem define
uma série de características dos aminoácidos, tais como polaridade, grau de
ionização em solução aquosa, cargas elétricas e o tamanho (DEVILIN, M.
Thomas. 2007. P.75-80).
Essa cadeia lateral é o que difere os aminoácidos em sua estrutura,
tamanho, cargas elétricas e solubilidade em água (polar ou apolar). Além de
conferir propriedades físico-químicas diferentes a cada aminoácido, os
resíduos laterais são também responsáveis por forças estabilizadoras,
advindas de interações fracas (ligações de hidrogênio, hidrofóbicas,
eletrostáticas etc.), que mantêm as estruturas conformacionais enoveladas das
proteínas (DEVILIN, M. Thomas. 2007. P.75-80).

6
O enovelamento de uma proteína é um processo fisioquímico através do
qual a estrutura de uma proteína assume a sua configuração funcional, a
organização espacial da proteína é resultado do tipo de aminoácido que a
compõe e de como eles estão dispostos uns em relação aos outros. Dessa
forma, o enovelamento é um processo físico-químico que leva a molécula de
um estado não enovelado à sua configuração de menor energia livre sendo
esse estado um mínimo local ou global (NOLASCO, 2010).
3.6. Cn-AMP1
O peptídeo Cn-AMP1 é um peptídeo curto com apenas nove resíduos de
aminoácidos, possui características catiônicas, hidrofóbicas e toxidade seletiva,
mas não possui nenhuma ação hemolítica. Extraído da água de coco verde
exerce atividade antimicrobiana contra bactérias gram-positivas e gram-
negativas (SILVA, 2012).
3.7. Mecanismos de ação dos peptídeos antimicrobianos
Os peptídeos antimicrobianos têm como principais características a
natureza catiônica e a capacidade de permeabilizar membranas de
microrganismos (Izadpanah&Gallo, 2005). Os peptídeos antimicrobianos são
proteínas de baixo peso molecular, menos de cem aminoácidos básicos,
possuindo atividade antimicrobiana contra bactérias, vírus e fungos, que lhes
oferecem uma carga liquida positiva. Alem disso, apresentam uma região rica
Figura 2: representação da estrutura
alfa hélice de uma proteína.
Figura 3: Representação da estrutura
folha beta de uma proteína.

7
em aminoácidos hidrofóbicos. Esses peptídeos isolados, geralmente
apresentam-se carregados positivamente e são na maioria das vezes,
anfipáticos, possuindo tanto domínio hidrofóbico quanto hidrofílico, o que
capacita a molécula a ser solúvel em ambiente aquoso e também em
membranas lipídicas (Izadpanah&Gallo, 2005).
A separação espacial entre aminoácidos básicos e hidrofóbicos torna os
peptídeos antimicrobianos moléculas anfipáticas. Muitos peptídeos
antimicrobianos ligam-se as membranas negativamente carregadas
permeabilizando-as, o que resulta na formação de uma via para a
movimentação de íons, soluto e do próprio peptídeo (McElhaney and Prenner,
1999). A ação dos antimicrobianos induz a defeitos na membrana, como
separação de fase ou afinamento da membrana, formação de poros ou
rompimento da bicamada lipídica (Lohner&Prenner, 1999).
Existem peptídeos que assumem uma estrutura anfipática de alfa hélice
quando se ligam a uma membrana. Sabe-se que a natureza hidrofóbica e
catiônica dos peptídeos é importante para interação ideal entre o peptídeo e a
membrana bacteriana (Hancock &Chappler, 1999).
Em geral os peptídeos são divididos em dois grandes grupos. Peptídeo
antimicrobiano linear são peptídeos que em solução aquosa ou que mimetize a
membrana celular, costumam adotar uma estrutura em hélice anfipática. Já os
peptídeos antimicrobianos cíclicos são peptídeos que podem apresentar as
suas extremidades amino e carboxi-terminal abertas ou fechadas e podem
formar estruturas tais como grampos tipo ou uma mistura de hélice e folhas
(BULET et al.,2004)
Os mecanismos de ação dos peptídeos antimicrobianos ainda não são
totalmente conhecidos, porém, sabe-se que seu caráter catiônico e a sua
tendência a anfipaticidade facilitam sua interação com a superfície celular de
bactérias e inserção na membrana (BROGDEN, 2005; ZASLOFF, 2002). As
interações eletrostáticas entre peptídeos antimicrobianos e superfícies
celulares de bactérias são facilitadas pela presença de fosfolipídios com carga
liquida negativa na face externa da membrana, grupos fosfatos de moléculas
de lipopolissacarídeos e ácidos teicóicos (BROGDEN, 2005; ZASLOFF, 2002).
A membrana citoplasmática de células de mamíferos, ao contrário das
bactérias, apresenta na sua face externa uma predominância de fosfolipídios

8
com carga liquida neutra, o que contribui para a ação de diversos peptídeos
antimicrobianos seja seletiva para membranas de bactérias (BROGDEN, 2005;
ZASLOFF, 2002). Como já mencionado, a anfipaticidade dos peptídeos
antimicrobianos facilita sua inserção na membrana por meio da interação de
sua região hidrofóbica com a região hidrofóbica dos fosfolipídios da membrana
e com consequência, pode ocorrer permeabilização da membrana, vazamento
do conteúdo intracelular e morte do patógeno (BROGDEN, 2005; HANCOCK;
SAHL, 2006; YOUNT et al., 2007).
O mecanismo de ação de peptídeos antimicrobianos é hipotético e se
baseia em três etapas: ligação de peptídeo em forma monomérica à membrana
plasmática, inserção de peptídeos na membrana para a formação de poros,
recrutamento progressivo de monômero para o aumento do diâmetro do poro
(NOLASCO, 2010). Para ilustrar a inserção na membrana e os danos
resultantes causados à célula, três modelos são propostos (NOLASCO, 2010).
3.8. Modelo carpete
A membrana é completamente coberta pela proteína. Quando uma
concentração critica é atingida, as proteínas provocam sérios danos à célula.
As proteínas permanecem na região de interface das membranas, em
conformação paralela, não entrando em contato com a cauda dos fosfolipídios.
O processo de desintegração e formação de micelas leva à morte da bactéria
(NOLASCO, 2010).
3.9. Modelo barril
Neste modelo, os peptídeos antimicrobianos anfipáticos em hélice
depois da interação eletrostática com a parte externa da membrana bacteriana,
formam poros do tipo barril, nos quais a porção hidrofóbica da proteína interage
com a porção hidrofóbica dos fosfolipídios da membrana e a região hidrofílica
da proteína fica voltada para dentro do poro. O vazamento do conteúdo
intracelular através destes poros pode levar à morte celular (NOLASCO, 2010).
3.10. Modelo dos poros toroidais ou canais agregados
Depois da interação com os fosfolipídios da membrana, várias moléculas
de peptídeo se juntam e formam um complexo com moléculas de água
9
associadas. Este complexo induz a produção de canais que atravessam a
membrana celular, sendo estes canais temporários, que podem permitir a
formação de íons, moléculas de grande massa molecular e, inclusive, do
próprio peptídeo, sem que haja grandes alterações na estrutura da membrana.
A diferença entre este modelo e o modelo barril é que os peptídeos estão
sempre associados com as cabeças polares dos fosfolipídios, mesmo quando
inseridos perpendicularmente à bicamada lipídica (NOLASCO, 2010).
4. Resultados e discussões
Nesta parte do trabalho foi feita uma analise dos gráficos gerados a
partir dos dados colhidos nas duas simulações, os primeiros quatro gráficos
farão alusão à simulação realizada com água TFE e o peptídeo, logo em
seguida os próximos gráficos representaram a segunda simulação contendo
somente água e o peptídeo.
VOLUME
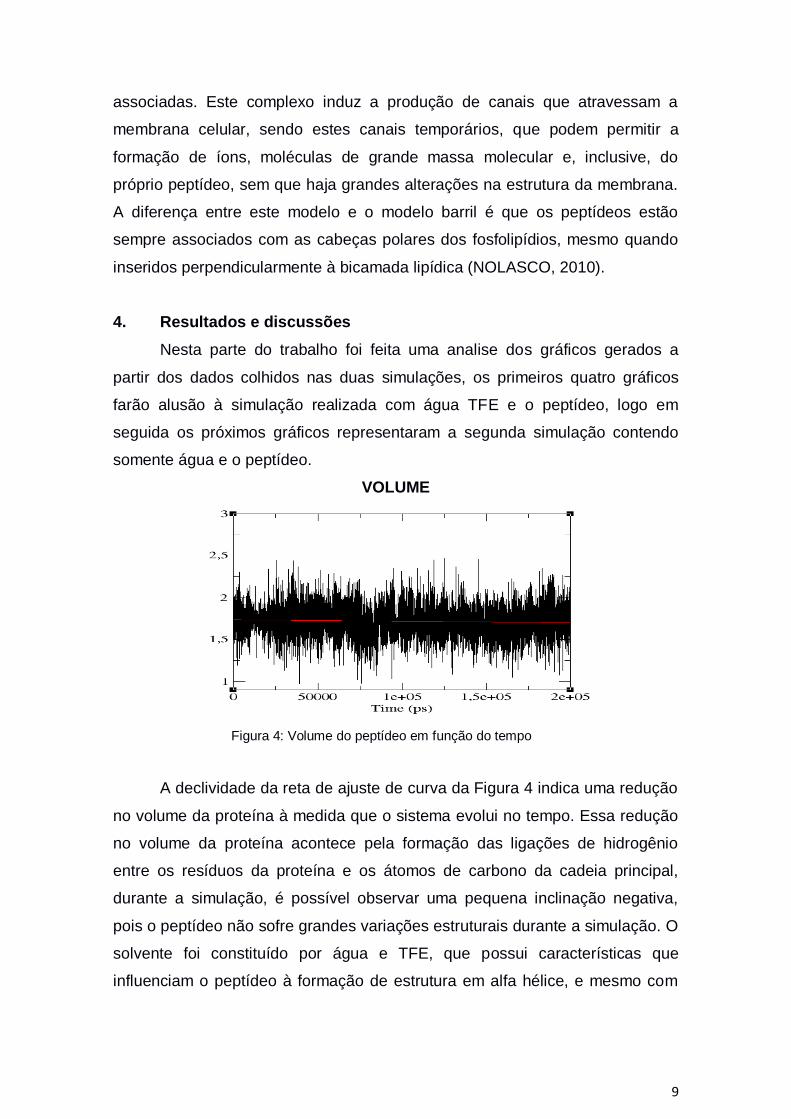
Figura 4: Volume do peptídeo em função do tempo
A declividade da reta de ajuste de curva da Figura 4 indica uma redução
no volume da proteína à medida que o sistema evolui no tempo. Essa redução
no volume da proteína acontece pela formação das ligações de hidrogênio
entre os resíduos da proteína e os átomos de carbono da cadeia principal,
durante a simulação, é possível observar uma pequena inclinação negativa,
pois o peptídeo não sofre grandes variações estruturais durante a simulação. O
solvente foi constituído por água e TFE, que possui características que
influenciam o peptídeo à formação de estrutura em alfa hélice, e mesmo com
10
um indutor desta estrutura (alfa hélice), foi possível observar que o peptídeo
não adotou estrutura alguma.
ÁREA
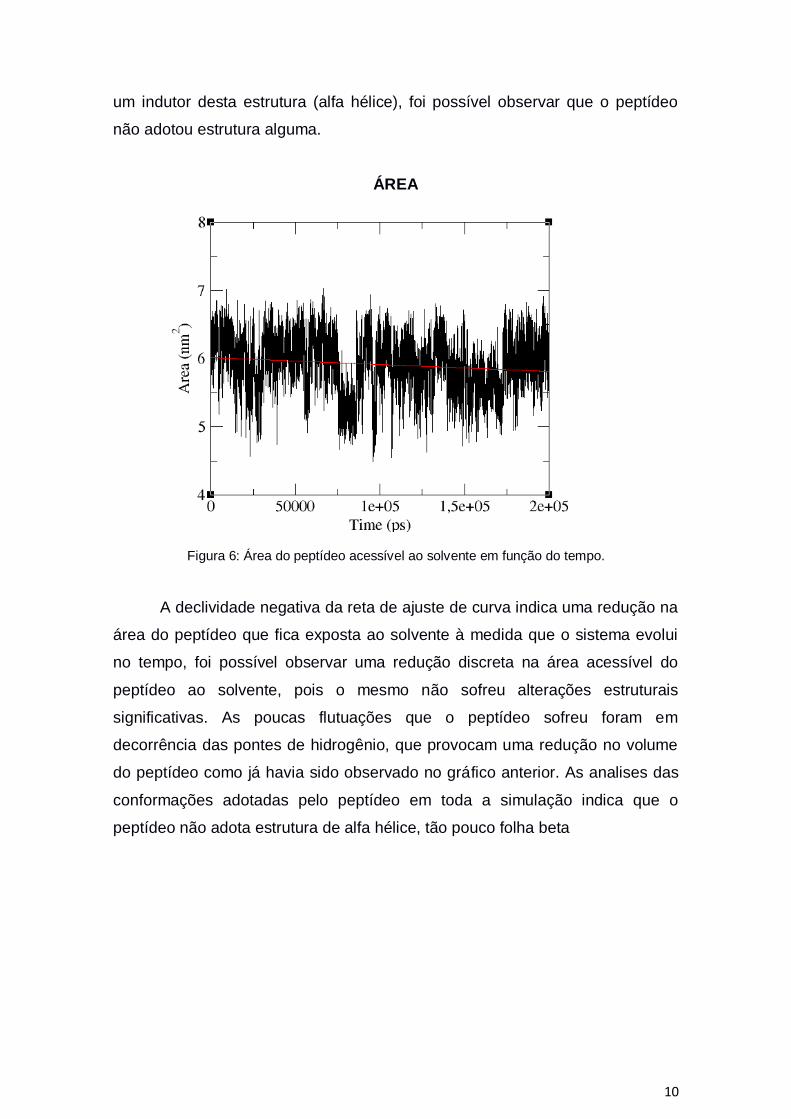
Figura 6: Área do peptídeo acessível ao solvente em função do tempo.
A declividade negativa da reta de ajuste de curva indica uma redução na
área do peptídeo que fica exposta ao solvente à medida que o sistema evolui
no tempo, foi possível observar uma redução discreta na área acessível do
peptídeo ao solvente, pois o mesmo não sofreu alterações estruturais
significativas. As poucas flutuações que o peptídeo sofreu foram em
decorrência das pontes de hidrogênio, que provocam uma redução no volume
do peptídeo como já havia sido observado no gráfico anterior. As analises das
conformações adotadas pelo peptídeo em toda a simulação indica que o
peptídeo não adota estrutura de alfa hélice, tão pouco folha beta
11
RAIO DE GIRO
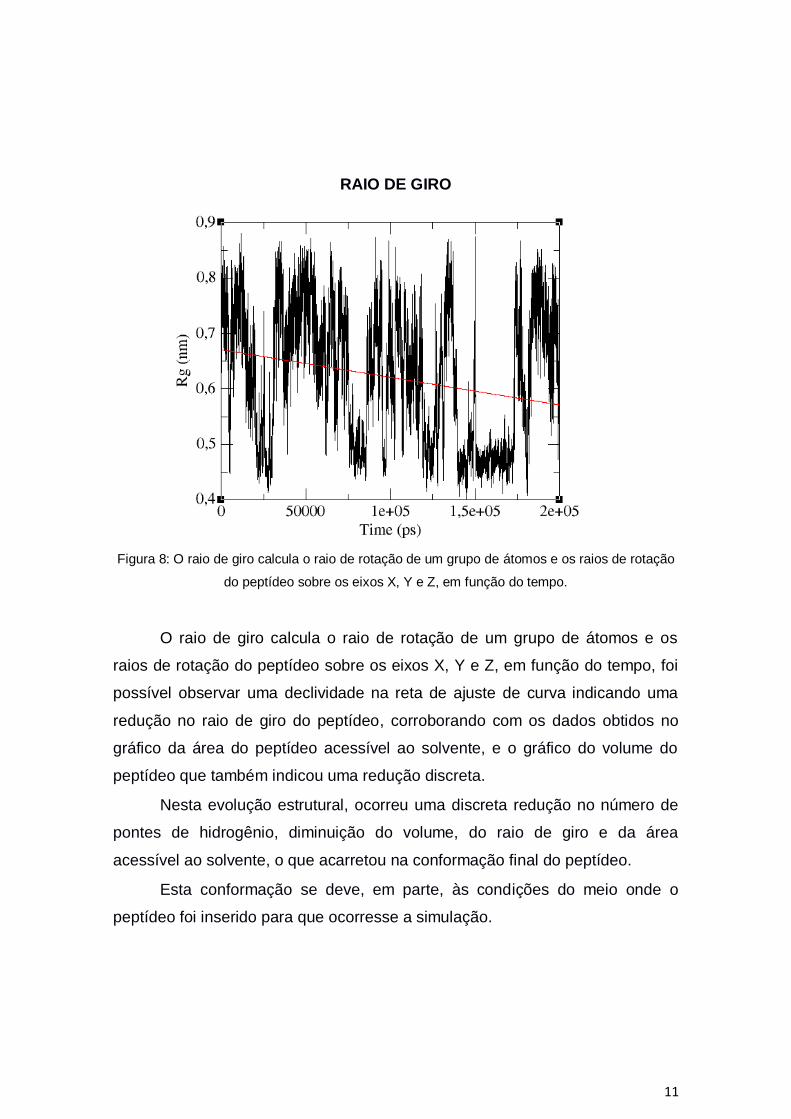
Figura 8: O raio de giro calcula o raio de rotação de um grupo de átomos e os raios de rotação
do peptídeo sobre os eixos X, Y e Z, em função do tempo.
O raio de giro calcula o raio de rotação de um grupo de átomos e os
raios de rotação do peptídeo sobre os eixos X, Y e Z, em função do tempo, foi
possível observar uma declividade na reta de ajuste de curva indicando uma
redução no raio de giro do peptídeo, corroborando com os dados obtidos no
gráfico da área do peptídeo acessível ao solvente, e o gráfico do volume do
peptídeo que também indicou uma redução discreta.
Nesta evolução estrutural, ocorreu uma discreta redução no número de
pontes de hidrogênio, diminuição do volume, do raio de giro e da área
acessível ao solvente, o que acarretou na conformação final do peptídeo.
Esta conformação se deve, em parte, às condições do meio onde o
peptídeo foi inserido para que ocorresse a simulação.
12
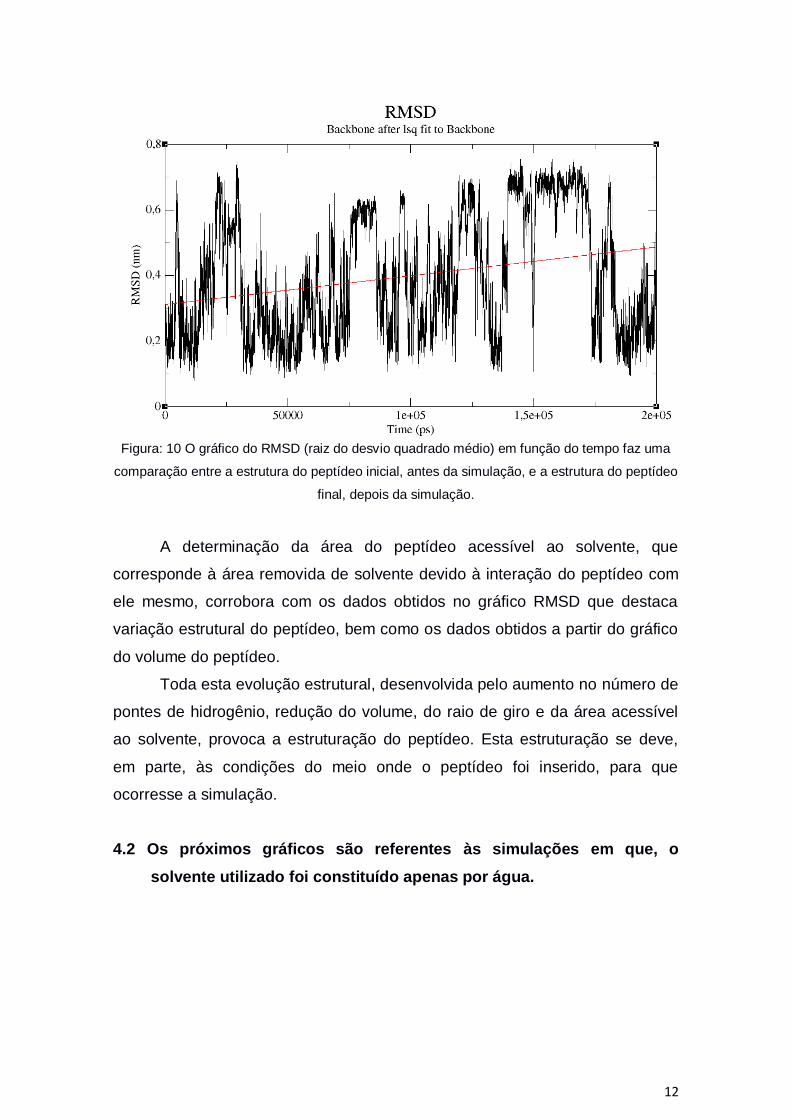
Figura: 10 O gráfico do RMSD (raiz do desvio quadrado médio) em função do tempo faz uma
comparação entre a estrutura do peptídeo inicial, antes da simulação, e a estrutura do peptídeo
final, depois da simulação.
A determinação da área do peptídeo acessível ao solvente, que
corresponde à área removida de solvente devido à interação do peptídeo com
ele mesmo, corrobora com os dados obtidos no gráfico RMSD que destaca
variação estrutural do peptídeo, bem como os dados obtidos a partir do gráfico
do volume do peptídeo.
Toda esta evolução estrutural, desenvolvida pelo aumento no número de
pontes de hidrogênio, redução do volume, do raio de giro e da área acessível
ao solvente, provoca a estruturação do peptídeo. Esta estruturação se deve,
em parte, às condições do meio onde o peptídeo foi inserido, para que
ocorresse a simulação.
4.2 Os próximos gráficos são referentes às simulações em que, o
solvente utilizado foi constituído apenas por água.
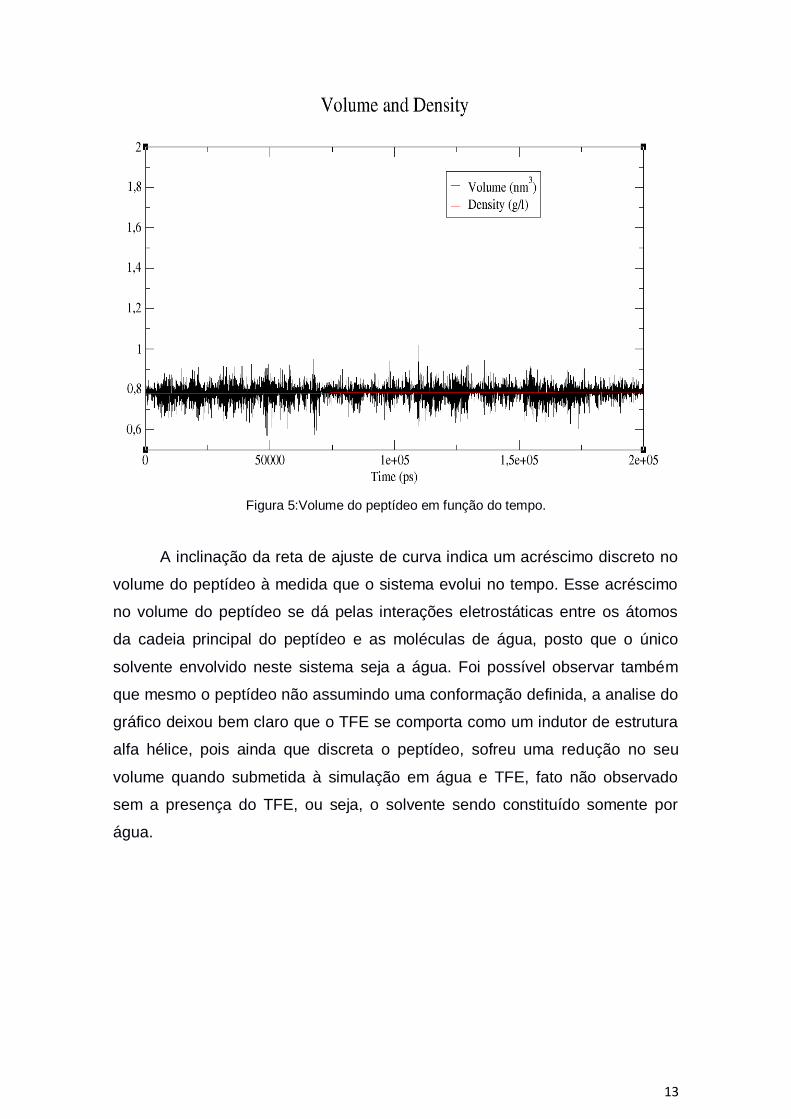
13
Figura 5:Volume do peptídeo em função do tempo.
A inclinação da reta de ajuste de curva indica um acréscimo discreto no
volume do peptídeo à medida que o sistema evolui no tempo. Esse acréscimo
no volume do peptídeo se dá pelas interações eletrostáticas entre os átomos
da cadeia principal do peptídeo e as moléculas de água, posto que o único
solvente envolvido neste sistema seja a água. Foi possível observar também
que mesmo o peptídeo não assumindo uma conformação definida, a analise do
gráfico deixou bem claro que o TFE se comporta como um indutor de estrutura
alfa hélice, pois ainda que discreta o peptídeo, sofreu uma redução no seu
volume quando submetida à simulação em água e TFE, fato não observado
sem a presença do TFE, ou seja, o solvente sendo constituído somente por
água.
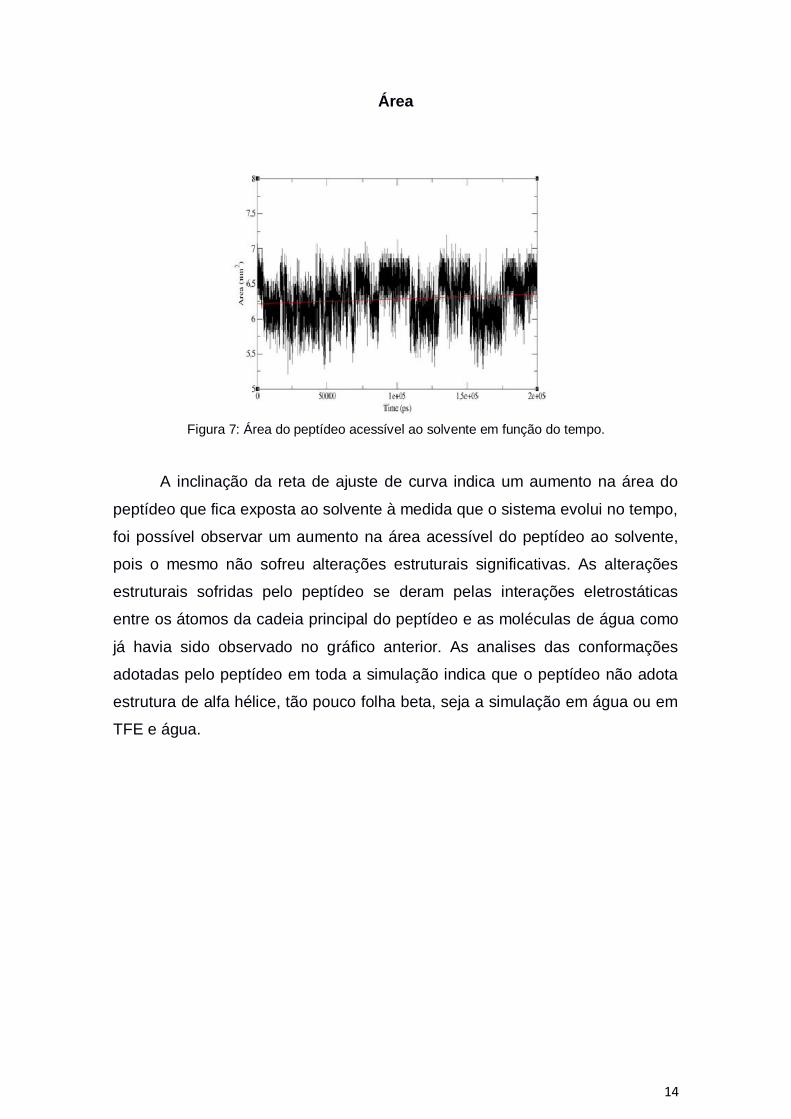
14
Área
Figura 7: Área do peptídeo acessível ao solvente em função do tempo.
A inclinação da reta de ajuste de curva indica um aumento na área do
peptídeo que fica exposta ao solvente à medida que o sistema evolui no tempo,
foi possível observar um aumento na área acessível do peptídeo ao solvente,
pois o mesmo não sofreu alterações estruturais significativas. As alterações
estruturais sofridas pelo peptídeo se deram pelas interações eletrostáticas
entre os átomos da cadeia principal do peptídeo e as moléculas de água como
já havia sido observado no gráfico anterior. As analises das conformações
adotadas pelo peptídeo em toda a simulação indica que o peptídeo não adota
estrutura de alfa hélice, tão pouco folha beta, seja a simulação em água ou em
TFE e água.
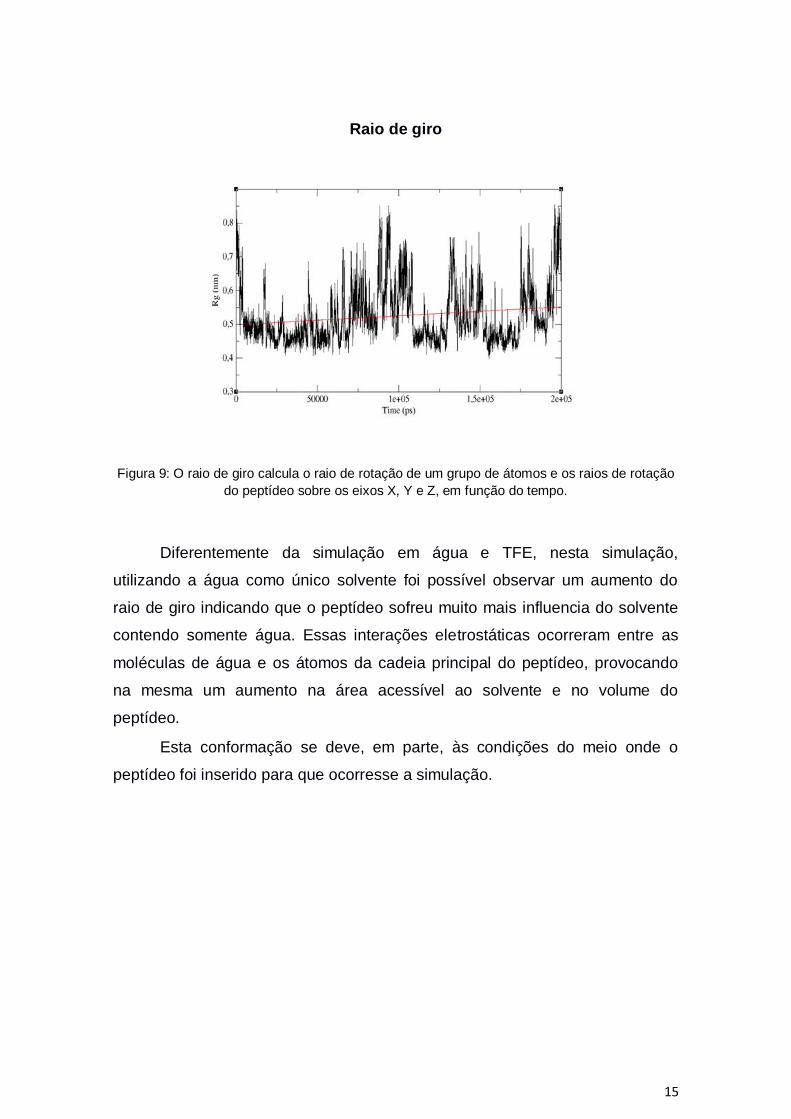
15
Raio de giro
Figura 9: O raio de giro calcula o raio de rotação de um grupo de átomos e os raios de rotação
do peptídeo sobre os eixos X, Y e Z, em função do tempo.
Diferentemente da simulação em água e TFE, nesta simulação,
utilizando a água como único solvente foi possível observar um aumento do
raio de giro indicando que o peptídeo sofreu muito mais influencia do solvente
contendo somente água. Essas interações eletrostáticas ocorreram entre as
moléculas de água e os átomos da cadeia principal do peptídeo, provocando
na mesma um aumento na área acessível ao solvente e no volume do
peptídeo.
Esta conformação se deve, em parte, às condições do meio onde o
peptídeo foi inserido para que ocorresse a simulação.
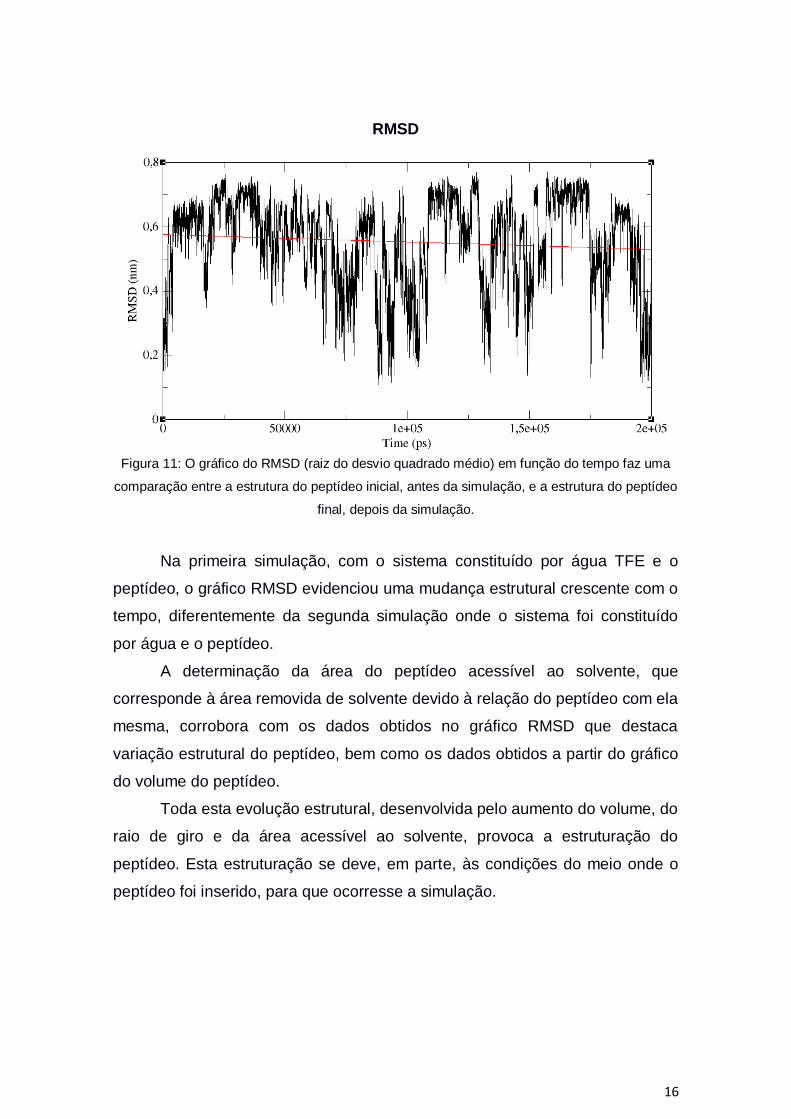
16
RMSD
Figura 11: O gráfico do RMSD (raiz do desvio quadrado médio) em função do tempo faz uma
comparação entre a estrutura do peptídeo inicial, antes da simulação, e a estrutura do peptídeo
final, depois da simulação.
Na primeira simulação, com o sistema constituído por água TFE e o
peptídeo, o gráfico RMSD evidenciou uma mudança estrutural crescente com o
tempo, diferentemente da segunda simulação onde o sistema foi constituído
por água e o peptídeo.
A determinação da área do peptídeo acessível ao solvente, que
corresponde à área removida de solvente devido à relação do peptídeo com ela
mesma, corrobora com os dados obtidos no gráfico RMSD que destaca
variação estrutural do peptídeo, bem como os dados obtidos a partir do gráfico
do volume do peptídeo.
Toda esta evolução estrutural, desenvolvida pelo aumento do volume, do
raio de giro e da área acessível ao solvente, provoca a estruturação do
peptídeo. Esta estruturação se deve, em parte, às condições do meio onde o
peptídeo foi inserido, para que ocorresse a simulação.

17
5. Conclusão
Diante de todos os dados colhidos nas duas simulações realizadas e
todas as análises elaboradas, a partir dos dados obtidos nas simulações, a
primeira simulação utilizando água e TFE como solvente e a segunda
simulação utilizando somente água como solvente foi possível apresentar a
análise conformacional do peptídeo CN-AMP1. O sistema foi analisado por
meio de simulação por dinâmica molecular.
A simulação por dinâmica molecular teve por objetivo a varredura do
espaço conformacional, no intuito de obter uma estatística que favorecesse a
proposição da estrutura tridimensional do peptídeo CN-AMP1. Os dados das
simulações permitem os seguintes comentários:
1. A tendência à conformação helicoidal do peptídeo não foi verificada e, a
partir desta apreciação, é possível inferir que a estruturação da
macromolécula é um processo energeticamente desfavorável.
2. A instabilidade de qualquer conformação, verificada ao longo da
trajetória, possibilita a proposição de que o peptídeo CN-AMP1 não
adota estrutura diferente da estrutura aleatória.
6. Referencias bibliográficas
BROGDEN, K. A. Antimicrobial peptides: pore formers or metabolic inhibitors in
bacteria?.Naturereviews. Microbiology, v. 3, n. 3, p. 238-50, 2005. Disponível em:
<http://www.ncbi.nlm.nih.gov/pubmed/15703760>. .
BULET, PHILIPPE; STÖCKLIN, R.; MENIN, L. Anti-microbial peptides: from
invertebrates to vertebrates. Immunological Reviews, v. 198, n. 1, p. 169-184,
2004.John Wiley \&
Sons.Disponívelem:<http://www.ncbi.nlm.nih.gov/pubmed/15199962>. .
CURTIS, Helena. Biologia. Rio de janeiro: Guanabara kooganS.A. , 1997. P.72-
73. )

18
DEVILIN, M. Thomas. Manual de Bioquímica com correlações clínicas. São
Paulo: Edgard Blucher. 2007. P.75-80
GAVIRA, M. D. O. SIMULAÇÃO COMPUTACIONAL COMO UMA
CONHECIMENTO. ,2003.
HANCOCK, R.E.W..; CHAPPLE, D.S. Peptides Antibiotics. Antimicrobial Agents and
chemotherapy, Washington, v.43, n.6, p.1317-1323, jun 1999.
HANCOCK, R. E. W.; SAHL, HANS-GEORG.Antimicrobial and host-defense
peptides as new anti-infective therapeutic strategies.NatureBiotechnology, v. 24, n. 12,
p. 1551-1557, 2006. Disponível em:
<http://www.ncbi.nlm.nih.gov/pubmed/17160061>. .
IZADPANAH, A .;GALLO, R.L. Antimicrobialpeptides. Jurnal of the American
Academy of Dermatology, St. Loius, v.52, n.3 pt1, p318-390, mar 2005.
LOHNER, K.; PRENNER, E.J. Diffrential scanning calorimetry and x-ray diffraction
studies of the specificity of the interaction of antimicrobial peptides with membrane-
mimic systems. Biochimica et BiophysicaActa, Amsterdam, v.1462, n.1-2,p.141-156,
dec 1999.
MCELHANEY, R. N.; PRENNER , E. J. The interaction antimicrobial peptides with
mode lipid bilayer and biological membranes.Biochimica et BiophysicaActa,
Netherlands, v.1462,p1-234,1999
NAMBA, A. M.; SILVA, V. B.; SILVA, C. H. T. P. Dinâmica molecular : teoria e
aplicações em planejamento de fármacos. , v. 33, p. 13-23, 2008.
NOLASCO, D. O. Estudos Estruturais por Dinâmica Molecular do Peptídeo Polybia-
mpi via Replica Exchange. ,2010.

19
ROSE, E.; APARECIDA, M. Estudo Estrutural de Polímeros – II - Dinâmica do
Polietileno de 100 Unidades de Repetição ( PE100 ), a Diferentes Temperaturas . , p. 1-
14, 1992.
SILVA, O. N. Biopolymers : Peptide Science CnAMP-1 : a new promiscuous peptide
with potential for microbial infections treatment Biopolymers : Peptide Science. ,2012.
Um panorama desse problema de saúde pública no Brasil. ., 2010.
YOUNT, N. Y.; WARING, ALAN J; GANK, K. D., et al. Structural correlates of
antimicrobial efficacy in IL-8 and related human
kinocidins.BiochimicaetBiophysicaActa, v. 1768, n. 3, p. 598-608, 2007.
ZASLOFF, MICHAEL. Antimicrobial peptides of multicellular organisms.Nature, v.
415, n. 6870, p. 389-395, 2002.NaturePublishingGroup. Disponível em:
<http://www.ncbi.nlm.nih.gov/pubmed/11807545>. .


















