
A. Insulina e outros fármacos utilizados no tratamento da … · A insulina interage com o RI...
71
Química Farmacêutica – PFFH – UP7 1 A. Insulina e outros fármacos utilizados no tratamento da Diabetes mellitus 1. Insulina - Conhecer estrutura, características, biossíntese e metabolismo. A insulina é uma hormona composta por 21 AA na cadeia A e 30 na cadeia B que estão ligadas por duas pontes dissulfureto. Separadamente, as cadeias são inactivas. Biossíntese 1. Sintetizada como pré-pró-insulina nas células β. 2. Translocação através da membrana do retículo endoplasmático rugoso (RER). Durante este processo, ocorre a clivagem de 24 AA do N-terminal da cadeia B -> Pró-insulina (t 1 / 2 = 17 min, mais solúvel que a insulina e alguma actividade insulin-like). 3. Dentro do RER, a proteína é dobrada para se formarem as 3 pontes dissulfureto. 4. No complexo de Golgi, a pró-insulina sofre uma modificação adicional catalisada por endopeptidases dependentes de Ca 2+ . Neste processo, 4 AA básicos e o peptídeo C são removidos por proteólise -> Insulina (t 1 / 2 = 5-6 min). No pâncreas, é armazenada como hexâmero -> Contém dois Zn 2+ e é constituído por 3 dímeros. Estes estão ligados primariamente por 4 pontes de H e uma interacção hidrofóbica envolvendo folhas-β, e por outras interacções. Receptor de Insulina (RI) É uma glicoproteína composta por dois heterodímeros e cada um deles é constituído por uma unidade α (extracelular, onde se liga a insulina) e outra β (transmembranar e com actividade tirosina-cinase). Após ligação da insulina à unidade α -> Autofosforilação de tirosinas das unidades β -> Fosforilação de proteínas intracelulares, que servem como sinais intracelulares. A insulina interage com o RI através de AA do N- e C-terminal da cadeia A e do C- terminal da cadeia B, ligando-se a AA localizados do N- e C-terminal da unidade α do RI, que inclui uma região rica em cisteína. Metabolismo (ocorre primariamente no fígado e rim) Após secreção, 50% chega ao fígado pela veia porta -> Clivagem das pontes dissulfureto (glutationa insulina transhidrogenase – insulinase) -> Degradação proteolítica -> Entrada na corrente sanguínea. A insulina é filtrada pelo glomérulo -> Reabsorção/degradação nos túbulos. Nos tecidos, a degradação na superfície celular é limitada.
Transcript of A. Insulina e outros fármacos utilizados no tratamento da … · A insulina interage com o RI...

Química Farmacêutica – PFFH – UP7
1
A. Insulina e outros fármacos utilizados no tratamento da Diabetes mellitus
1. Insulina
- Conhecer estrutura, características, biossíntese e metabolismo.
A insulina é uma hormona composta por 21 AA na cadeia
A e 30 na cadeia B que estão ligadas por duas pontes
dissulfureto. Separadamente, as cadeias são inactivas.
Biossíntese
1. Sintetizada como pré-pró-insulina nas células β.
2. Translocação através da membrana do retículo
endoplasmático rugoso (RER). Durante este processo,
ocorre a clivagem de 24 AA do N-terminal da cadeia B ->
Pró-insulina (t1/2 = 17 min, mais solúvel que a insulina e
alguma actividade insulin-like).
3. Dentro do RER, a proteína é dobrada para se formarem as 3 pontes dissulfureto.
4. No complexo de Golgi, a pró-insulina sofre uma modificação adicional catalisada por
endopeptidases dependentes de Ca2+. Neste processo, 4 AA básicos e o peptídeo C são
removidos por proteólise -> Insulina (t1/2 = 5-6 min). No pâncreas, é armazenada como
hexâmero -> Contém dois Zn2+ e é constituído por 3 dímeros. Estes estão ligados
primariamente por 4 pontes de H e uma interacção hidrofóbica envolvendo folhas-β, e
por outras interacções.
Receptor de Insulina (RI)
É uma glicoproteína composta por dois heterodímeros e cada um deles é constituído
por uma unidade α (extracelular, onde se liga a insulina) e outra β (transmembranar e com
actividade tirosina-cinase). Após ligação da insulina à unidade α -> Autofosforilação de
tirosinas das unidades β -> Fosforilação de proteínas
intracelulares, que servem como sinais intracelulares.
A insulina interage com o RI através de AA do N- e C-terminal da cadeia A e do C-
terminal da cadeia B, ligando-se a AA localizados do N- e C-terminal da unidade α do RI, que
inclui uma região rica em cisteína.
Metabolismo (ocorre primariamente no fígado e rim)
Após secreção, 50% chega ao fígado pela veia porta -> Clivagem das pontes
dissulfureto (glutationa insulina transhidrogenase – insulinase) -> Degradação proteolítica ->
Entrada na corrente sanguínea. A insulina é filtrada pelo glomérulo -> Reabsorção/degradação
nos túbulos. Nos tecidos, a degradação na superfície celular é limitada.

Química Farmacêutica – PFFH – UP7
2
- Conhecer as diferentes insulinas utilizadas no tratamento da diabetes mellitus, incluindo as
suas estruturas, principais características, a obtenção e a estabilidade/metabolismo.
De forma a controlar os níveis glicémicos,
inicialmente os diabéticos apenas tinham como alternativas
viáveis a insulina bovina ou porcina pois, apesar das
diferenças, a bovina difere em 3 AA e a porcina apenas num
-> Problema: desenvolvimento de anticorpos -> Duas vias de síntese para a insulina humana:
1) Síntese das cadeias separadamente, seguida de ligação destas -> Hormona activa;
2) Fermentação para sintetizar o péptido da hormona -> Isolamento da substância pura.
Posteriormente surgiram as técnicas de DNA recombinante (usadas actualmente)
para produzir insulina -> Inserção de genes em E. coli seguida de métodos fermentativos.
Estrutura da insulina em solução
Em solução, a insulina pode existir como monómero, dímero ou hexâmero. Contudo,
só é activa como monómero (apenas este consegue interage com RI) -> Dissociação dos
hexâmeros/dímeros após administração. Este monómero existe nas baixas [] fisiológicas, mas
a [] elevadas (Ex.: formulações farmacêuticas) a insulina dimeriza e, a pH neutro na presença
de Zn2+ forma hexâmeros -> Alterações na sua [] pode alterar a absorção por via subcutânea.
A estrutura secundária e terciária é muito semelhante, mas a estrutura primária tem
pequenas diferenças entre espécies. A cadeia A tem duas hélices-α e a B tem uma hélice-α e
uma folha-β -> Resíduos hidrofóbicos da cadeia A estejam no interior do péptido -> Promove
solubilidade em água e estabilidade. A presença de fenol e cresol (fenol com CH3 em o, m ou p)
muito usados para preservar formulações de insulina, produz mudanças nesta conformação.
Estabilidade da insulina
Os Zn2+ são importantes para estabilizar as preparações de insulina. Actualmente,
todas são soluções de zinco ou suspensões de formas insolúveis de zinco com insulina.
A preparação de longa-duração e mais estável é a insulina zinco-protamina ->
Precipitação da insulina na presença de Zn2+ e protamina (proteína básica). Contém 2 Zn2+ por
hexâmero de insulina. Uma insulina de menor duração de acção e mais útil utiliza protamina
neutra Hagedorn (NPH), que inclui m-cresol (conservante) – insulina isofânica. Seis m-cresol
estão associados ao hexâmero.
Posteriormente percebeu-se que a adição de Zn2+ a preparações hexaméricas com
dois Zn2+ em tampão acetato permitia que a insulina cristalizasse sob várias formas (com
diferente dissolução em água). Neste sentido, a insulina amorfa rapidamente solúvel é uma
forma com 2 Zn2+ e a cristalina, com solubilidade mais lenta, é uma forma com 4 Zn2+.

Química Farmacêutica – PFFH – UP7
3
Uma forma adicional de insulina corresponde àquela que é parcialmente unfolded.
Esta forma precipitados viscosos e insolúveis (fibrilhas) -> O shielding dos domínios
hidrofóbicos é o factor principal para a agregação. Quando estes domínios expostos (A2, A3,
B11, and B15) interagem com os resíduos alifáticos mais interiores (A13, B6, B14, and B18) no
hexâmero, formam-se fibrilhas. A sua formação é acelerada quando a insulina está como
monómero; e não resuspendem se a preparação for agitada -> Farmaceuticamente inactivas.
Quanto à instabilidade química, a forma solúvel rápida de insulina era uma solução
com zinco com pH 2-3. Esta, se armazenada a 40C (derivado desaminado constitui 6-12% da
proteína após 6 meses) -> C-terminal da Asn, em condições ácidas, sofre ciclização a anidrido.
Este pode reagir com a água -> Desamidação; ou com o N-terminal
da Phe de outra cadeia -> Molécula com ligações cruzadas. Se
armazenada a 250C, o derivado desamidado constitui ~90% da
proteína após 6 meses.
Insulina armazenada a pH neutro -> Desamidação na Asn
B3 -> Insulinas com aspartato e isoaspartato têm actividade igual
à da insulina nativa; clivagem da cadeia entre Thr A8 e Ser A9 e
formação de ligações cruzadas covalentes com uma 2ª cadeia de
insulina ou com protamina (se presente). As duas últimas situações
são processos lentos quando comparados com desamidações, mas
dão origem a produtos que causam reacções alérgicas.
Factores que afectam a duração de acção da insulina
Análogos da insulina
Alterações/Remoções de AA do C-terminal
da cadeia B de insulina não alteram muito a
actividade, mas podem influenciar a velocidade de
formação/separação do dímero.
Estas modificações permitem obter vários
tipos de insulina. Assim sendo, as preparações desta podem ser divididas em 4 grupos:
- Acção ultra-curta, com muito rápido início de acção (Ex.: lispro) -> Na insulina lispro, os
resíduos 28 e 29 foram trocados -> Não afecta interacção com RI, mas reduz a tendência para a

Química Farmacêutica – PFFH – UP7
4
insulina se organizar em hexâmeros (comuns na insulina humana e mais insolúveis que
monómeros). Já na insulina Aspart, a troca de AA tem o mesmo efeito.
- Acção curta (Ex.: regular) -> São dispensadas com a insulina em solução, o que permite um
rápido início de acção. Também se encontram preparações que consistem em insulinas de
acção curta misturadas com insulinas de acção intermédia -> Controlar aspectos PK.
- Acção intermédia (Ex.: NPH ou isofânica, lente, insulina-zinco) ou longa, com início lento de
acção (Ex.: ultralente) -> Ambas são dispensadas como suspensões túrbidas, com mobilização
retardada da insulina do local de injecção -> Criação de complexos fracamente hidrossolúveis
da insulina aniónica com protamina (policatiónica) em tampão fosfato (insulina NPH) ou com
zinco em tampão acetato (insulinas lente ou ultralente). A insulina glargina foi o 1º análogo de
longa duração, pois as modificações proporcionaram um ponto isoeléctrico ~7 -> Precipitação
após administração subcutânea e consequente libertação lenta do depot. Outro análogo é a
insulina detemir, que resulta da adição da N-acilação da Lys B19 com ácido mirístico. Este
ácido gordo liga-se à albumina, criando um depot. Tem duração inferior à da anterior.
- Conhecer os principais aspectos da REA das insulinas.
A actividade biológica da insulina (e a formação de hexâmeros e dímeros) reside mais
na estrutura espacial da molécula que em AA específicos -> Os últimos 3 AA da cadeia B
podem ser removidos sem perda de actividade, mas a clivagem do C-terminal da cadeia A
(Asn-21) leva à perda total da actividade. De modo geral, pode ocorrer substituição de AA
desde que não ocorram alterações na geometria global da molécula.
2. Desenvolvimento de fármacos úteis no tratamento da Diabetes mellitus relacionados com
outras hormonas que não a insulina.
- Conhecer a estrutura, principais características e obtenção do glucagon, GLP-1 e amilina, e
moléculas que interferem nas suas vias e podem ser úteis.
Glucagon
É um polipéptido formado nas células α do
pâncreas que eleva os níveis de glucose. É biossíntetizado
como pró-glucagon que é clivado para produzir glicentina que,
por sua vez, é clivada a glucagon. O glucagon humano é
comercializado para o tratamento de overdose de insulina.
É possível que terapias que o bloqueiem tenham
efeito significativo na gestão da diabetes mellitus tipo 2 ->
Pesquisa de moduladores do receptor de glucagon não
peptídicos -> Agonistas/antagonistas moléculas da família das

Química Farmacêutica – PFFH – UP7
5
quinoxalinas, acilhidrazidas e pirimidonas.
Glucagon-like peptide-1 (GLP-1)
É uma incretina, uma hormona peptídica natural secretada (no intestino delgado) em
resposta à ingestão de alimentos -> Estimula libertação de insulina e inibe a de glucagon. É
formado por dois péptidos (um com 30 AA, outro com 31). O GLP-1 por si só teria potencial
farmacológico, se não fosse o t1/2 de 90s. A dipeptidil peptidase IV (DPP-IV), encontrada nos
capilares intestinais e fígado, metaboliza-o: remove 2 AA do N-terminal -> Péptido resultante
tem t1/2 de 2 min. A administração de GLP-1 melhorou a função das células β, mas como não é
aconselhado a sua administração contínua -> Agonistas de GLP-1 e inibidores da DPP-IV.
Agonistas do GLP-1
Nos anos 90’s, descobriu-se que a saliva de
lagartos (Gila monsters) continha um péptido (exendin-
4) com actividade glucoreguladora. Este liga-se ao receptor de GLP-1 e mimetiza a sua
actividade, sendo a sua versão sintética a exenatida -> Partilha 53% de homologia de AA com o
GLP-1, é metabolizada pela peptidase, tem t1/2 de 2-4h, após administração subcutânea.
Amilina
É um péptido normalmente co-secretado com a insulina, das
células β em resposta a refeições. Diminui a libertação de glucagon e o
esvaziamento gástrico. Produz os efeitos por ligação a receptores
específicos no SNC, mas a administração é impraticável devido às suas
propriedades físico-químicas (péptido é insolúvel e agrega em solução)
-> Fármacos relacionados.
Acetato de Pramlintida -> Agonista da amilina
- Análogo da amilina no qual a Pro substituiu os AA das posições 25, 28 e 29 -> Aumentou
solubilidade aquosa e reduziu a tendência de auto-agregação -> Permite via subcutânea.
- Formulada a pH 4 -> Incompatível com insulina (pH 7.8) se administrados na mesma seringa.
3. Agentes hipoglicemiantes orais
3.1 – Sulfonilureias
- Compreender o seu desenvolvimento e conhecer as estruturas e principais características e
relacioná-las com a sua farmacocinética.
Os anti-diabéticos orais (ADO’s) – moléculas orgânicas pequenas – são fármacos que
baixam os níveis sanguíneos de glucose, substituindo a acção da insulina. São divididos em:
o Secretagogos de insulina (sulfonilureias de 1ª e 2ª geração e meglitinidas);
o Biguanidas (Ex.: metformina, fenformina);

Química Farmacêutica – PFFH – UP7
6
o Tiazolidinadionas (Ex.: rosiglitazona, pioglitazona);
o Inibidores de α-glucosidades (Ex.: acarbose, miglitol).
O desenvolvimento das sulfonilureias partiu de sulfonamidas antibacterianas, o IPTD
– 2-(p-aminobenzenosulfonamida)-5-isopropiltiadiazole – usado no tratamento de febre
tifóide, mas provocava morte por ser um agente hipoglicémico.
Através de modificações estruturais, obteve-se a
carbutamida (1º agente hipoglicémico sulfonilureia), contudo
foi retirada pelos seus efeitos na medula óssea e pelo desenvolvimento de
resistências bacterianas devido à sua acção anti-bacteriana -> A substituição
do NH2 por CH3 originou a tolbutamida, que manteve a acção hipoglicemiante,
mas já não tem acção antibacteriana.
As sulfonilureias podem ser divididas em 1ª geração (tolbutamida, tolazamida,
aceto-hexamida e cloropropamida) e 2ª geração (gliburida, glipizida e glimepirida), sendo que
os fármacos da 2ª tendencialmente têm t1/2 superiores e menos efeitos secundários.
1ª geração
- Cloropropamida: tem o maior t1/2 das sulfonilureias (32 h) ->
grande tendência para efeitos adversos. Uma explicação pode
ser o seu metabolismo lento (hidroxilação ω e ω-1 do propilo),
com ~20% a ser excretado na forma inalterada.
- Tolbutamida e tolazamida: sofrem oxidação benzílica mais
rápida -> Derivado ácido benzóico inactivo. A hidroxilação
alternativa do anel alifático da tolazamida a um metabolito
activo -> Acção mais prolongada em relação à tolbutamida.
- Aceto-hexamida: redução da C=O a OH (principal metabolito
2,5x mais activo); e hidroxilação do ciclohexilo em 4’ (inactivo).
2ª geração
- Glipizida e gliburida: metabolismo extenso -> Metabolitos menos
activos/inactivos excretados na
urina ou igualmente na urina e
bílis, respectivamente.
- Glimepirida: metabolizada pelo
CYP2C9 a M-1 (activo) e depois a
M-2 (inactivo).
As sulfonilureias têm elevada
ligação proteica (albumina) e, como

Química Farmacêutica – PFFH – UP7
7
tal, um grande volume de distribuição. São metabolizadas no fígado e os metabolitos são
excretados por via renal. Por último, a comida retarda a sua absorção, mas não afecta a
biodisponibilidade.
Estes fármacos ligam-se a canais de K+ dependentes de ATP (bloqueados ao
aumentar a relação ATP/ADP como consequência do metabolismo da glucose), que são
dímeros formados por uma subunidade α e uma β, também designada SUR (Sulfonyl urea
receptor). Após ligação -> Inibição do efluxo de K+ -> Despolarização -> Abertura de canais de
Ca -> Influxo de Ca2+ -> Produção e libertação de insulina.
- Conhecer a REA dos fármacos deste grupo.
As sulfonilureias são ácidos fracos com Pka ~5.
Sulfonilureia normal: anel aromático
monosubstituido, (geralmente para) com um substituinte alifático volumoso no N da ureia.
Grupos alquilo pequenos, (Ex.: metilo ou etilo) -> Não são activos.
NH2 da anilina: não-substituído ou com um substituinte que
seja removido in vivo.
X: pode ser O, S ou N incorporado numa estrutura
heteroaromática (Ex.: tiadiazol, pirimidina) ou acíclica (Ex.: ureia ou
tioreia). No caso das ureias, o N2 deve ter uma cadeia com pelo
menos 2 carbonos.
Na 1ª geração, o substituinte aromático é um átomo simples
ou então CH3, NH2, acetil, Cl, Br, CH3S, trifluorometil. Já na 2ª geração,
os grupos são maiores -> Potência significativamente maior. A
glimepiridina (2ª geração) apresenta um grupo de ligação similar ao
da gliburida e glipizida, no entanto, a diferença no seu perfil
farmacológico pode justificar uma classificação diferente.
3.2. Meglitinidas – conhecer as estruturas e principais características
Repaglinida (secretagogo de insulina)
- Tal como a glibenclamida e glimepirida, tem conformação em U -> Grupos hidrofóbicos
cíclicos no fim e uma ligação peptídica no fundo -> Análogos da repaglinida inactivos têm
conformação diferente e maior distância entre os grupos hidrofóbicos cíclicos.
Nateglinide (secretagogo de insulina)
- Principais metabolitos: derivados hidroxilados -> Conjugação com glucoronido.

Química Farmacêutica – PFFH – UP7
8
3.3. Biguanidas – compreender o seu desenvolvimento e conhecer as estruturas e principais
características e relacioná-las com a sua farmacocinética.
Historicamente, goat’s rue era usada como remédio tradicional para a diabetes ->
Descobriu-se que o princípio activo da planta, a galegina (isoamilenoguanidina), era tóxico ->
Descoberta de que a guanidina baixava os níveis de glucose em animais, mas era demasiado
tóxica para ser usada -> Fenformina (com actividade anti-diabética, mas provocava morte por
acidose láctica) -> Metformina (segura e com actividade anti-diabética).
Metformina: rapidamente absorvida no intestino, com biodisponibilidade entre 50-
60%. Não está ligada às proteínas, tem um elevado volume de
distribuição e acumula-se na parede do intestino, criando um
depot, que ajuda a manter as [] plasmáticas. É excretada na
urina (secreção tubular), sem sofrer metabolismo e tem t1/2 = 2-5h.
3.4. Tiazolidinadionas - Conhecer a sua descoberta, as estruturas e a sua farmacocinética.
Também conhecidas por “glitazonas”, são
exemplificados pela ciglitazona (1ª glitazona), cujos efeitos
hipoglicémicos foram descobertos por acaso.
O primeiro fármaco desta classe a ser comercializado
foi a troglitazona (aparentava ter poucos efeitos adversos, mas
mais tarde foram detectados casos de falência hepática,
necessidade de transplantes de fígado e morte) -> Descoberta de duas
novas glitazonas: rosiglitazona e pioglitazona.
As tiazolidinadionas diferem pela natureza dos grupos
ligados ao núcleo 2,4-tiazolidinediona. São extensamente
metabolizadas -> Todas as alterações metabólicas ocorrem no 2º arilo
ou adjacente a este.
Quanto à troglitazona, parece que a toxicidade hepática
pode estar associada a um dos seus metabolitos, nomeadamente M-3
(quinona – muito reactivo). O principal metabolito em humanos é M-1
(conjugação com sulfato).
3.5. Inibidores de α-glucosidades
- Compreender o metabolismo de hidratos de carbono complexos.
Para serem absorvidos do TGI para a corrente sanguínea, os
hidratos de carbono complexos ingeridos (Ex.: amido) devem primeiro
ser hidrolisados a monossacarídeos -> Os inibidores da α-glucosidase

Química Farmacêutica – PFFH – UP7
9
(Iα-g) impedem a hidrólise, podendo reduzir a taxa de absorção dos hidratos de carbono.
Amido: digerido pela amilases salivares e pancreáticas -> Dissacarídeos (maltose),
trissacarídeos (maltotriose) e oligossacarídeos (dextrina) -> Hidrólise a monossacarídeos pelas
oligosacaridases. Estas estão localizadas nas vilosidades do intestino delgado e são
constituídas por duas classes:
o -galactosidases: hidrolisam os -dissacarídeos (Ex.: lactose);
o -glucosidases: actuam nos -dissacarídeos (Ex.: maltose, isomaltose, sacarose).
- Conhecer as estruturas, principais características e a REA dos fármacos deste grupo.
Farmacóforo: compreende um ciclohexano substituído e um
anel 4,6-didesoxi-4-amino-D-glicose (conhecido como carvosina) -> A
amina secundária deste núcleo impede um grupo carboxilo da α-
glucosidase de protonar as ligações glicosídicas de oxigénio do substrato.
Têm sido desenvolvidos Iα-g que se assemelham a
aminoaçúcares simples, tais como miglitol e voglibose.
Parece que os Iα-g não bloqueiam a hidrólise dos dissacarídeos,
mas sim atrasam-na. A acarbose é pouco absorvida para a corrente
sanguínea, logo não se encontra associada a toxicidade sistémica. No
intestino, é degradada por amílases e bactérias, com alguns dos produtos a serem absorvidos
sistemicamente e excretados na urina.
B. Adrenocorticóides
1. Nomenclatura e estrutura geral dos esteróides
- Principais características estruturais destes compostos
- Aspectos gerais da nomenclatura deste grupo de moléculas
Esteróides -> 4 anéis fundidos (A, B, C e D), sendo hidrocarbonetos ciclopentano-
peridrofenantrenos (núcleo fenantreno reduzido,
saturado, ligado a um ciclopentano).
Colestano -> Esteróide com 27C, cadeia lateral de 8C,
ligados na posição 17.
Numeração: inicia em A e B até C10, depois para C, em C11 e acaba em C17 no D. Os metilos
angulares (para fora do plano) são numerados 18 (ligado a C13) e 19 (ao C10). A cadeia lateral
ligada a C17, começa em C20 e prossegue.
Com uma representação planar, a estrutura esteróide torna-se um plano com duas
superfícies: β (para cima – linhas normais) e α (baixo -
tracejado). Ex.: 5α -> configuração do H em C5, que é oposta do

Química Farmacêutica – PFFH – UP7
10
C19 do metilo angular -> Configuração trans entre A e B. Também há fusão trans entre B/C e
C/D.
As cadeias laterais em 17 são sempre β, a menos que indicadas a tracejado ou na
nomenclatura (Ex.: 17β).
Os esteróides podem ser desenhados em conformação em cadeira, e os anéis devem ser
equatoriais uns em relação aos outros. Se os metilos angulares em 18 e 19 forem β e forem
perpendiculares ao plano (axiais), as restantes orientações podem ser atribuídas. Ex.: No
5α-colestano, o C19 do CH3 ligado em 10, é sempre β-axial, e as 2 ligações em 3 devem ser β-
equatoriais e α-axiais.
A orientação das restantes ligações no esteróide podem ser determinadas se recordarmos que
os grupos no ciclohexano, posicionados nos C adjacentes (vicinais, -C1H-C2H- ) do anel (isto é,
1,2 um em relação ao outro) são trans se a sua relação é 1,2-diaxial ou 1,2- diequatorial e são
cis se a sua relação for 1,2-equatorial-axial.
O 5α colestano tem os anéis A/B, B/C e C/D fundidos em trans -> Esqueleto trans-anti-trans-
anti-trans; o 5β colestano tem um esqueleto cis-anti-trans-anti-trans: A e B em cis.
O termo syn é usado de modo semelhante a anti para definir uma relação do tipo cis, e não
existem esteróides naturais com a geometria do tipo syn.
A maneira convencional de desenhar os esteróides não mostra os H em 8β, 9α ou 14β, e os
metilos em C18 e C19 são desenhados com linhas sólidas.
Quase todos os esteróides bioactivos têm um esqueleto do tipo colestano. Mas dentro
dos activos também há: 5α-pregnanos (21C’s), 5α-androstanos (19C’s) e 5α-estranos
(18C’s). Adrenocorticóides -> Pregnanos (Ex.: Cortisona-> 17α,21-dihidroxipregn-4-eno-
3,11,20-triona).
Geralmente existe dupla em 4 e 5 ou 5 e 6 -> Retira geometria cis/trans entre A e B.
O símbolo Δ -> dupla ligação carbono-carbono num esteróide:
- Se se situar em 4 e 5 -> Referido como um Δ4-esteróide.
- Se se situar em 5 e 10 -> Δ5(10)-esteróide.
O colesterol (colest-5-en-3α-ol) é um Δ5-esteróide, ou mais especificamente, um Δ5-
esterol, visto ser um álcool insaturado.
Acetato de cortisona -> 17α,21-dihidroxipregn-4 eno-3,11,20-triona 21 acetato. A
progesterona, (pregn-4-eno-3,20-diona) também é análogo pregnano. Androgénios
também baseados no 5α-androstano, ex.: Testosterona (17β-hidroxiandrost-4-en-3-ona)
Desidroepiandrosterona (DHEA) -> Maior androgénio adrenal, denomina-se 3β-
hidroxiandrost-5-en-17-ona - androstano.

Química Farmacêutica – PFFH – UP7
11
Os estrogénios são análogos estranos, cujo anel A é aromático. O 17β-estradiol é o estra-
1,3,5,(10)-trieno-3,17β-diol. As cadeias alifáticas em 17 assumem-se β, quando aplicada
anomenclatura do colestano ou pregnano -> Notação 17β só necessária quando se encontrar
em α (17α).
- Diferentes séries de esteróides e características estruturais diferenciadoras
-conhecer a estrutura básica dos glucocorticóides e mineralocorticóides
Cortisona -> Glucocorticóide. Moléculas com 21C’s, derivadas da pregnenolona, um pregnano,
com cetona em C3. Em C11 pode-se encontrar 1 hidroxilo (cortisol) ou cetona (na cortisona).
Frudrocortisona-> Mineralocorticóide. 21C’s, com um OH em c11 e uma cetona em c3.
- Obtenção dos esteróides mais representativos a partir da diosgenina e do estigmasterol
Diosgenina (vinda das dioscoreas) + anidrido acético a
200ºC na autoclave Diacetato.
Oxidação crómica ataca quimiosselectivamente a dupla
da cadeia lateral-> Cetoéster tratado com ácido acético
a acetato de 16-desidropregnolona.
Redução catalítica quimioselectiva-> Acetato de
pregnolona-> Progesterona, por hidrólise alcalina e
oxidação de Oppenauer:
-Reacção reversível na qual o álcool 2º, por
tratamento do alcóxido de Al (em geral,
isopropóxido) e excesso de cetona que serve como
aceitador de H proveniente do álcool (ciclohexanona
ou acetona).
Usada na oxidação de álcoois esteroidicos.
Ao oxidar-se um esterol C3 e, como consequência do
meio básico da reacção, forma-se isomerização da
dupla em 5,6 (se existir) à posição conjungada 4,5.
A 16-desidropregnolona é precursora da

Química Farmacêutica – PFFH – UP7
12
desidroepiandrosterona (DHE), chave na síntese de testosterona (TRT), dos androgénios e
anabolizantes derivados desta, da estrona e estradiol.
Por redução de Birch -> 19-noresteróides anovulatórios. A transformação de 16-
desidropregnolona em DHE dá-se por transposição de Beckamann, da sua oxima-> N-
acetilamina correspondente. A hidrólise desta-> Acetato de DHE e a hidrólise posterior seguida
da reacção de Oppenauer -> Androstenediona.
Para evitar a exploração excessiva de
dioscóreas Concebido o processo de
Upjohnque: Usa o estigmasterol (esterol
abundante não insaponificável do azeite de
soja) como matéria-prima.
Isolar o estigmasterol de impurezas: Lixiviação.
Oxidação de Oppenauer, seguida da
degradação da cadeia C17 até conversão à cadeia de progesterona Possível por ozonólise
redutora para dar o 20-formil derivado, posteriormente convertido em enamina em C22 que
se oxida em várias condições (ex: Ozonização e fotooxigenação) para dar progesterona.
Síntese de esteróides com anel aromático
Esteróide mais simples: Estrogénio em que o anel A é aromático.
Plantas com esteróides em quantidades consideráveis, não possuem estruturas com o anel A
aromático Obtidos por semi-síntese de outros esteróides cujo passo prévio é a aromatização
do Anel A. Para isto, elimina-se o CH3 angular em C10 que é o C19.
Estratégias da androstenodiona: Androsterona, por tratamento com Br em ácido acético ou
NBS Derivado dibromado.
Reacção com colidina (trimetilpiridina) 1,4-dienona.
Pirólise em azeite vegetal a 600ºC Estrona
(rendimento < 30%).
Modificação mais recente: Carbonilo em C17 da
dienona é protegido com propilencetal e o A é
aromatizado por redução com lítio-bifenilo. Em meio
ácido o produto hidrolisa também o cetal Estrona
com rendimento de 60%. Redução desta com hidretos metálicos Estradiol.
Reacções com esteróides: Existe ataque estereosselectivo na face (menos impedida pela
existência de CH3 angulares e (possível) cadeia C17).

Química Farmacêutica – PFFH – UP7
13
Etinil e seu éster metílico (mestranol), componentes dos contraceptivos orais: preparam-se por
estinilação da estrona e do seu éter metílico com acetato potássico em amoníaco líquido.
Produtos são os que se formaram por ataque na face .
4.2. Biossíntese e metabolismo
- Formação da pregnenolona a partir do colesterol.
Glândulas adrenais: colesterol -> Clivagem enzimática da cadeia lateral a
pregnenolona (3β- hidroxipregn-5-en-20-ona) -> Precursor biossintético dos
adrenocorticóides.
Biotransformação: executada por citocromo P450. 3 passos de oxidação
envolvidos na conversão; são consumidos 3 moles de NADPH e O2 por cada mol de
colesterol convertido a pregnenolona:
1ª oxidação -> formação de colest-5-ene-3β,22R-diol (passo a);
2ª oxidação -> produção de colest-5-ene-3β,20R,22R-triol (passo b);
3ª oxidação -> Clivagem da ligação C20-C22 -> libertação de pregnenolona e aldeído
isocapróico (passo c).
- Formação de glucocorticóides e mineralocorticóides a partir da pregnenolona.
Biossíntese destes é regulada por mecanismos independentes.
Glucocorticóides (cortisol) -> Influência de hormonas peptídicas secretadas pelo hipotálamo e
hipófise anterior (adenohipófise) que activam o córtex adrenal (eixo HPA).
Mineralocorticóides (corticosterona e aldosterona) -> Influência da
angiotensina II -> Animais hipofisectomizados: Formação de aldosterona
apenas levemente diminuída ou inalterada. O balanço electrolítico
permanece quase normal.
A hormona peptídica na pituitária anterior que influencia a biossíntese de
corticosteroides é a ACTH (corticotropina) e no hipotálamo é o factor de
libertação de corticotropina (CRF):
Produção de ambas: Regulada pelo SNC por feedback negativo corticóide.
CRF é libertado pelo hipotálamo -> pituitária anterior -> Estimula a
libertação de ACTH para a corrente sanguínea. Esta é transportada para as
glândulas adrenais-> Estimula a biossíntese e secreção dos glucocorticóides.
Os níveis circulantes de glucocorticóides agem no hipotálamo e pituitária anterior para regular
a libertação de CRF e ACTH.
Pregnenolona: convertida a adrenocorticóides por série de oxidações enzimáticas e
isomerização da dupla ligação.

Química Farmacêutica – PFFH – UP7
14
Passos seguintes da biossíntese-> No retículo endoplasmático da célula de córtex adrenal:
Hidroxilação da pregnenolona em 17 pela enzima 17α-hidroxilase (CYP17)-> 17α-
hidroxipregnenolona (passo b).17α-hidroxilo: Acção hormonal dos adrenocorticóides.
Num só passo, a 17α-hidroxipregnenolona é oxidada a um intermediário 3-ceto pela acção da
enzima 5-ene-3β-hidroxiesteróide desidrogenase (3β-HSD) e isomerizada a 17α-
hidroxiprogesterona pela enzima 3-oxoesteróide-4,5-isomerase (passos c e d).
Outra hidroxilação ocorre por acção da 21-hidroxilase (CYP21)-> 11-desoxicortisol, que contém
a cadeia lateral cetol (-COCH2OH) fisiologicamente importante em 17β (passo e). A ausência
da CYP21 impede a biossíntese de cortisol, desviando o excesso de 17α-hidroxipregnenolona e
17α-hidroxiprogesterona para a superprodução de andrógenos C19.
Passo final: 11β-hidroxilase (CYP11B2)-> Hidrocortisona (cortisol, passo f), o glucocorticóide
endógeno mais potente secretado pelo córtex adrenal.
Via para a formação do potente mineralocorticóide aldosterona-> semelhante à da
hidrocortisona, usa várias das mesmas enzimas.
Via preferida: Conversão de pregnenolona a progesterona pela 5-ene-3β-hidroxiesteróide
desidrogenase e a 3-oxoesteróide-4,5-isomerase (passos c e d).
Hidroxilação em C21 da progesterona pela 21-hidroxilase-> 21-hidroxiprogesterona (11-
desoxicorticosterona (passo e).
11β-hidroxilase (CYP11B2) catalisa a conversão da 21-hidroxiprogesterona a corticosterona
(passo f), que apresenta actividade mineralocorticóide.
As duas oxidações finais envolvem hidroxilações no metilo C18 e são catalisadas pela 18-
hidroxilase (passo g)-> 18-hidroxicorticosterona e depois aldosterona, a secreção
mineralocorticóide endógena mais potente do córtex adrenal.
O aldeído em C18 existe em equilíbrio com a sua forma hemiacetal.
- Principais aspectos do metabolismo dos corticosteróides.
Hidrocortisona (hormonalmente activa) e cortisona (metabolito inactivo da
hidrocortisona) -> Interconvertíveis pela 11β-hidroxiesteróide desidrogenase.
Presentes 2 isozimas da 11β-hidroxisteróide desidrogenase: tipo 1 (11β-
HSD1), isozima “do fígado”, e tipo 2 (11β-HSD2), isozima “do rim”.
Isozima 11β-HSD1: bidireccional, interconverte rapidamente a hidrocortisona
e a cortisona, encontrada em muitos tecidos. Importante na regulação da gluconeogénese
hepática e na produção de gordura nos tecidos adiposos.
Isozima 11β-HSD2: unidireccional, catalisa a 11β-desidrogenação da hidrocortisona para dar
cortisona. Placenta e rins, especificamente nos túbulos contornados distais e ductos colectores
retí
culo
end
op
lasm
átic
o
mit
ocô
nd
ria

Química Farmacêutica – PFFH – UP7
15
corticais. Importante no rápido metabolismo da hidrocortisona, impedindo assim a ligação
desta aos receptores de mineralocorticóides. Deficiência de 11β-HSD2-> Doenças genéticas
hereditárias, excesso de mineralocorticóides (hipertensão, retenção excessiva de sais e
hipocalemia por níveis elevados de hidrocortisona no rim).
Hidrocortisona: Metabolizada pelo fígado após administração por qualquer via. Excretada na
urina como conjugados O-glucurónido e O-sulfato de urocortisol. 5β-dihidrocortisol e
urocortisona (menores).
Tetrahidrourocortisol -> Principal met., geometria do 5β-pregnano e função 3α-hidroxilo. A
configuração 5β é semelhante à geometria anelar dos ácidos biliares não hormonais.
Todos os adrenocorticóides activos contêm cetona em 3 e dupla em 4,5. Formação de
metabolitos 5β da hidrocortisona:
Redução da dupla 4,5 a uma geometria 5β para os anéis A e B (cis) pela 5β-redutase ou
Redução da 3-cetona pela 3α-hidroxiesteróide desidrogenase (3α-hidroxilo) ou 3β-
hidroxiesteróide desidrogenase (3β-hidroxilo).
Estas reacções representam as principais vias metabólicas dos glucocorticóides e dos seus
homólogos endógenos. (Urocortisol -> cortisol, urocortisona -> cortisona)
Oxidação reversível do 11β-hidroxilo de glucocorticóides pela 11β-hidroxiesteróide
desidrogenase inactiva estes fármacos e limita a actividade mineralocorticóide. Outras vias:
6β-hidroxilação (CYP3A4) e redução da 20-cetona -> análogos 20-hidroxilo;
Oxidação da cadeia lateral 17-cetol a ácidos 17β-carboxílicos e perda da cadeia lateral 17-cetol
-> esteróides 11β-hidroxi-17-ceto-C19.
Alguns metabolitos adrenocorticóides anéis A aromáticos assemelham-se a estrogénios.
4.3. Interacção corticosteróides-receptor
- Estrutura do receptor glucocorticóides, Interacções e resíduos importantes na interacção.
O receptor de glucocorticóides: membro da superfamília de receptores nucleares. É uma
proteína solúvel encontrada no citosol. Domínio de ligação: 11 hélices α e 4 cadeias β. Bolsa de
ligação que distingue dos rec. de estrogénios, androgénios e mineralocorticóides.
Quando um glucocorticóide se liga ao receptor, é completamente incluso dentro de uma bolsa
formada pelas hélices 3, 4, 5, 6 e 7 e as cadeias β 1 e 2. A forte afinidade de ligação resulta de
interacções hidrofílicas e hidrofóbicas com resíduos AA. Contactos mais significativos: pontes
de H entre os grupos hidrofílicos nos glucocorticóides.
Carbonilo do anel A -> Pontes de H com Arg-611 e o amido de Gln-570.
Hidroxilos em C11 e C21 -> Pontes de H com a cadeia lateral de Asn-564.
Hidroxilo em C17 -> Ponte de H com Gln-642.

Química Farmacêutica – PFFH – UP7
16
Carbonilo C20 -> Ponte de H com Thr-739.
- Conhecer a REA da actividade glucocorticóide/mineralocorticóide.
Glucocorticóides naturais interagem com o receptor de mineralocorticóides.
Sintéticos Diminuição de efeitos mineralocorticóides, aumento do efeito AI glucocorticóide.
Muitos são preparados para melhorar os parâmetros FK Ésteres lipofílicos e hidrofílicos.
Grupos funcionais essenciais para a actividade mineralocorticóide e glucocorticóide:
o Esqueleto pregnano com uma ‘espinha dorsal’ trans total (B/C e C/D);
o Sistema de anéis A-en-ona (anel A ∆4-3-cetona);
o Cadeia lateral 17β -cetol (C20-ceto-C21-hidroxi, -COCH2OH).
O C21-OH deve estar livre para a actividade. Excepções: derivados C21 Cl e C20 fluorometilo
tio éster (-SCH2F), retêm actividade AI topicamente ou por inalação.
Os anéis C e D (11, 12, 13, 16, 17, 18, 20 e 21) são mais importantes para a ligação que A e B.
Substituições aumentam a actividade anti-inflamatória:
Achatamento do anel A Adição de dupla em C1 (Δ1) e de OH em C11.
Substituições Aumentam propriedades AI e mineralocorticóides:
Grupos 17α-hidroxi, 9α-flúor ou 9α-cloro.
Substituintes diminuem a actividade mineralocorticóide:
Grupos 16α- ou 16β-metilo, 16α-hidroxilo ou um 16α,17α-acetónido.
Substituintes específicos podem aumentar a actividade AI como diminuir as propriedades
mineralocorticóides, dependendo deste:
Um C6α-CH3 aumenta ligeiramente a actividade AI, e um grupo C6α-F duplica o efeito AI.
Ambos, em combinação com substituinte C16, diminuem muito os efeitos mineralocorticóides.
4.4. Desenvolvimento de fármacos corticosteróides
- Conhecer as três gerações de corticóides e suas características.
Primeira geração -> Cortisona, cortisol e corticosterona.
Cetonas α,β-insaturadas em C3, com uma cetona ou hidroxilo em C11 e uma cadeia
lateral de natureza 17α,21-dihidroxi-20-ona. Cortisona, in vivo -> Cortisol (hidrocortisona) e é
mais estável -> VO. Cortisol -> via parenteral.
Segunda geração -> Prednisona (Δ1-11-cetona) e prednisolona, seu análogo 11β-hidroxilado.
Nestes análogos, aumentou-se a potência sem aumentar efeitos mineralocorticóides
(alteração da geometria do anel A que supõe a incorporação de duplas adicionais). Introdução
de insaturações: aumenta a flexibilidade molecular. Os compostos insaturados em C1 e C4
podem existir como vários confórmeros: Diferenças energéticas mínimas.

Química Farmacêutica – PFFH – UP7
17
Regressão da actividade AI de uma série de derivados de cortisol com um substituinte em C9
(F, Cℓ, Br, I, OH, CH3, CH3O, OC2H5 e SCN)-> Actividade AI correlaciona-se com o efeito
indutor, o tamanho e o parâmetro π de solubilidade.
Actividade aumenta quando o substituinte na dita posição retira carga (σ positivo), diminui o
seu tamanho ou aumenta a hidrofobia. F-> Mais activo dos derivados halogenados.
Efeito da retirada da carga eléctrica-> Aumento da acidez do 11β-hidroxi-> ponte de H mais
forte entre este grupo e o receptor.
Fluoração em 9α (fludrocortisona) aumenta a actividade mineralocorticóide, pelo que estes
compostos se usam apenas topicamente com anti-inflamatórios.
Introdução simultânea de uma dupla e um metilo em C6-> 6α-metilprednisolona.
Introdução de um hidroxilo em C16α nos 9α-fluoroderivados-> triamcinolona e seu acetónido,
usado no tratamento de transtornos dermatológicos.
Outros: dexametasona, betametasona -> têm metilo em C16α ou C16β.
Terceira geração -> Deflazacort, um análogo da prednisolona com um anel de oxazolina
condensado em C16 e C17, que se comporta como um pró-fármaco.
Hidrolisado por esterases séricas ao 21-hidroxiderivado, metabolito activo ->
Efeitos quase só AI, sem efeitos mineralocorticóides. Sem
efeitos sobre o metabolismo do glucídico e do Ca (evita a
osteoporose e diabetes produzidas pelo tratamento
continuado).
RU26988 e RU28362: a cadeia de dihidroxiacetona em
C17 dos corticóides naturais foi substituída por um resto
17α-alquinil-17β-hidroxi.
Introdução de substituintes em C11β nestes-> derivados
que se ligam fortemente ao receptor de progesterona
(Ex.: mefepristona).
- Principais características físico-químicas dos corticosteróides;
- Estratégias utilizadas para modular a lipofilia/hidrofilia destes compostos e consequências.
- Estrutura com a via de administração, potência de acção e características FK.
Glucocorticóides: lipofílicos apesar de terem pelo menos três OH. Podem ser esterificados
com ácido apropriado-> Aumento ou diminuição da lipofilicidade.
OH C21: é o mais acessível -> Mais fácil de esterificar.
OH C17: ligeiramente impedido pela cadeia lateral C17 -> Reage mais lentamente.
OH C11: Altamente impedido pelos metilos em C10 e C13 -> Não reage com ácidos.

Química Farmacêutica – PFFH – UP7
18
Esterificação tem efeito na afinidade para o receptor e no metabolismo dos glucocorticóides.
OH C21 deve estar livre para formar uma ponte de H com Arg-564 no receptor de
glucocorticóides-> Ésteres C21: Pró-fármacos-> Hidrólise para se tornarem activos.
OH C17 tem de estar livre para ser oxidado metabolicamente-> Esterificação do
OH C17 inibe a oxidação ao grupo C17 ceto, prolongando a duração.
Os ésteres glucocorticóides lipofílicos não se concentram na urina (sofrem
reabsorção tubular após filtração glomerular) -> Duração de acção prolongada.
Lipofilia de um glucocorticóide pode ser aumentada por esterificação com ácidos lipofílicos:
Aumento do log P (actividade local, menor absorção sistémica e efeitos 2ºs diminuídos).
Pró-fármacos ésteres lipofílicos em C21-> Maior duração e início de acção mais lento (os
ésteres lipofílicos são volumosos e retardam as enzimas hidrolíticas). Administração:
Oral; por inalação -> tratamento da asma e da DPOC; tópica. Diésteres e cetais -> aumento
adicional do log P e da duração de acção (metabolismo lento).
Diésteres e hexacetónidos: pró-fármacos. Cetais planos não o são (têm C21-OH livre).
Diésteres são formados em C21 e em OH C17 ou C16, se presente. Os ésteres em C17 ou C16
são activos intactos.
Diésteres mais comuns: diacetato ou dipropionato.
Cetal: derivado cíclico formado entre hidroxilos em C adjacentes e um C carbonilo de cetona.
Mais comum: acetónido (condensação entre OH C16 e 17 e carbonilo da acetona
(CH3COCH3)).
Se o acetónido estiver junto a um éster C21-t-butilacetato-> hexacetónido, o “hex” refere o nº
de C encontrados no butilacetato.
Hexacetónidos são pró-fármacos-> Hidrólise para se tornarem activos.
Buteprato (probutato): diéster que consiste num propionato em C21 e um butirato em C17.
Pró-fármaco; o éster C21 deve ser hidrolisado.
Esterificação também usada para aumentar a hidrofilicidade-> Pró-fármacos solúveis em água.
Usa-se um ácido dicarboxílico ou ácido fosfórico para condensar com o OH C21-> coloca um
grupo ionizável (carboxilato/fosfato) no pró-fármaco->
Ésteres hidrosolúveis
Usados para preparar produtos injectáveis aquosos para
administração IM ou IV.
Pró-fármacos hidrofílicos: Rápido início de acção->
Hidrolisados pelas esterases do plasma.
Contrário aos pró-fármacos lipofílicos: Facilmente

Química Farmacêutica – PFFH – UP7
19
excretados no rim-> Acção curta.
Pró-fármacos hidrofílicos: mais efeitos sistémicos-> Ampla distribuição: Elevada solubilidade.
Podem ser injectados por via IV-> Emergências asmáticas (status asmático) -> Doente incapaz
de tomar medicação oral.
4.5. Agentes específicos – Estrutura e características dos principais corticosteróides
Glucocorticóides sistémicos
Hidrocortisona - Endógena.
- Actividade glucocorticóide e mineralocorticóide.
- Actividades glucocorticóide e mineralocorticóide são avaliadas por esta.
- Grupos essenciais para as actividades: esqueleto pregnano com ‘espinha
dorsal’ trans-total, o anel A-en-ona e a cadeia lateral 17β-cetol.
- A actividade glucocorticóide é aumentada pelos OH em C11 e 17.
- Disponível em várias formas éster.
Prednisolona
- Hidrocortisona à qual foi adicionada uma dupla Δ1-> Duas duplas no anel A, que o achatam e
aumentam a acção glucocorticóide em detrimento da mineralocorticóide.
- Maior duração de acção-> Dupla ligação extra no anel A retarda a redução
metabólica.
- Como álcool livre para VO. Éster fosfato de Na C21 disponível para uso parenteral.
- Vários pró-fármacos éster. Pró-fármaco prednisona: 11-ceto da prednisolona:
Convertido in vivo ao 11β-activo, necessário para a ponte de H a Asn-564 no GR.
- Não usada em doentes com disfunção hepática-> Podem não reduzir o 11-ceto.
Metilprednisolona
- Adição de 6α-metilo à prednisolona -> Aumento da act. glucocorticóide, abolição da
mineralocorticóide. Disponível como o álcool livre para VO e como vários ésteres.
Glucocorticóides para inalação:
Dipropionato de beclometasona
- Pró-fármaco lipofílico.
- 16β-metilo: Diminui actividade mineralocorticóide e o 9α-cloro aumenta as actividades
glucocorticóide e mineralocorticóide.
- Metabolizado ao mais activo 17α-monopropionato durante absorção nos pulmões e
adicionalmente metabolizado ao álcool livre no fígado.
- O dipropionato também é metabolizado ao 21-monopropionato inactivo no fígado.
Budesonido

Química Farmacêutica – PFFH – UP7
20
- Acetal formado entre os 16α,17α-dihidroxilo e butanal.
-Glucocorticóide não halogenado com acetal 16,17: Diminui actividade mineralocorticóide.
- Epímero R mais activo que o epímero S.
- O C21 OH está livre-> Não é pró-fármaco.
- Absorvida sistemicamente, elevada ligação e metabolizada a 16α-hidroxiprednisolona e 6β-
hidroxibudesonida no fígado.
Flunisolide
- Cetal acetona (acetonido) com um 6α-F e um OH livre em C-21.
- Acetonido :Diminui actividade mineralocorticóide. 6α-F: Aumenta actividade glucocorticóide.
- Não é pró-fármaco, porque tem o OH livre em C-21.
- 20% de afinidade aos receptores e 40% BD.
- Metabolizado pelo CYP3A4 ao 6β-hidroxi, com <1% da actividade do composto original.
Fluticasona
- Trifluorometilo único em C20. Em combinação com éster 17α-propionato->
Maior afinidade para o receptor de glucococorticóides.
- 9α-flúor -> aumenta as actividades glucocorticóide e mineralocorticóide.
- 6α-flúor -> aumenta apenas a acção glucocorticóide.
- Eficácia -> efeito local (e não sistémico). Dose inalada possui elevada ligação proteica.
- Não é pró-fármaco; Extensamente metabolizado no fígado.
- Único metabolito detectável -> Ácido 17β-carboxílico (oxidação pelo CYP3A4).
Furoato de mometasona
- Grupos únicos que conferem actividade melhorada e vantagens FK.
-Cl em C21 e éster ácido furóico em C17 -> Maior afinidade para o receptor de glucocorticóides
de qualquer corticosteroide tópico.
- 16α-metilo -> Diminuição dos efeitos mineralocorticóides.
- 9α-cloro -> Aumento das actividades glucocorticóide e mineralocorticóide.
- Acção local (menor BD sistémica dos glucocorticóides inalados).
- Extensamente metabolizado. Metabolitos 6β-hidroximetasona, 21-hidroximetasona e outros.
Acetonido de triancinolona
- Função acetonida, que diminui a actividade mineralocorticóide.
- O 9α-fluoro aumenta a actividade glucocorticóide e mineralocorticóide.
- Tem afinidade 10x mais que a beclometasona e 6x menos que a do furoato de mometasona.
- Metabolizado no fígado a: 6β-hidroxitriamcinolone acetonida e o C-21 carboxi-6β-
hidroxitriamcinolone acetonida, prontamente excretados pelos rins.

Química Farmacêutica – PFFH – UP7
21
- Acetonida: Intacta durante reacções metabólicas-> Resistente à clivagem hidrolítica.
- É excretado como metabolitos na urina (40%) e fezes (60%). <1% excretado inalterado.
Antagonistas adrenocorticóides – conhecer as estruturas e principais características
Competem para a ligação aos receptores esteróides (antiglucocorticóides ou
antimineralocorticóides) e inibidores da biossíntese de adrenosteróides. Os complexos
receptores-antagonistas não estimulam a produção de novos mRNA e proteínas.
Espironolactona e análogos ligam-se aos receptores mineralocorticóides no rim: Acção
diurética e hipotensiva
Essencial a acção do anel A 3-ceto-4eno para o antagonismo-> abertura diminui a actividade.
Difere dos glucocorticóides tendo um 18-aldeído (participa no equilíbrio tautomérico,
formando o hemiacetal), não tendo 17α-OH, com a sua lactona e 7-tiolester.
A progesterona também demonstra actividade anti-mineralocorticóide em []>10-4M.
Maiorias dos não esteróides inibem complexos do P450 envolvidos na biossíntese dos
adrenoesteróides. Metirapona: Reduz a biossíntese do cortisol por inibição da 11-β hidroxilase
mitocondrial. Usada para testar a função adrenal-hipofisária e a habilidade de secretar ACTH.
Aminoglutatimida inibe a clivagem da cadeia lateral e é usada como adrenalectomia médica.
Muitos antifúngicos azoles inibem a síntese de adrenocorticóides. Cetoconazole inibe a síntese
de esteróides fúngicos a baixas[]. A elevadas inibe a CYP3A4 na síntese de adrenoesteróides.
Trilostano: Inibidor esteróide da 3β-hidroxiesteroide desidrogenase usado no síndrome
de Cushing.
Bebida de Glycyrrhiza glabra produz intoxicação mineralocorticóide, pelo principio activo
àcido glicirrético e carbenoxolona (inibe a 11β-HSD) e a sua isoforma 2 no rim.
Os ácidos biliares como o quenodesoxicólico e litocólico inibem selectivamente a 11β-HSD1.
Úteis nos síndromas metabólicos para reduzir [] elevadas de glucose e como anti-diabéticos.
C. Funcionamento da tiróide e fármacos tiroideus
1. Introdução - Estrutura e principais características das hormonas da glândula
tiróide, bem como a sua biossíntese e conhecer e compreender a sua FK.
Hormonas da tiróide -> AA com iodo derivados da L-tirosina. T3 e T4 ->
Sintetizados na tiróide e armazenados como AA de tiroglobulina A T3 (3,5,3’-
triiodo-L-tironina ) é mais potente e uma parte é obtida da T4 (3,5,3’,5’-tetraiodo-
L-tironina) por deiodinases fora da tiróide.
A glândula tiróide contém também:
DIT (3,5-diiodo-L-tirosina)
MIT (3-iodo-L-tirosina)
T2 (3,3’-diiodo-L-
tironina)

Química Farmacêutica – PFFH – UP7
22
T3reversa(3,3’,5’-triiodo-L-tironina)
O acoplamento de dois anéis externos de DIT ou de um de DIT com um de MIT leva à formação
das duas hormonas principais da tiróide, T3 e T4, respectivamente.
Tiroglobulina:
1/3 do peso da glândula da tiróide; Matriz para a formação de T3 e T4;
Forma de armazenamento de hormonas e de iodo.
Biossíntese das hormonas da tiróide
Síntese de T3 e T4-> Regulada pela tirotropina (TSH). Esta estimula a síntese de
tiroglobulina, tiroperoxidase (TPO) e peróxido de hidrogénio. É necessária uma fonte exógena
de iodo que pelo impedimento estereoquímica tem um papel nas conformações de T3 e T4.
1) Transporte activo de iodeto pelas células foliculares
Aporte adequado de iodeto necessário para a síntese das hormonas.
O iodo da dieta é convertido em I- e quase completamente absorvido no TGI.
O mecanismo que permite à tiróide concentrar o I- sanguíneo contra o gradiente na célula
folicular é a bomba de iodeto (NIS: Na+/I- simporte) regulada pela TSH.
2) Oxidação do iodeto e formação das iodotirosinas
Para servir como iodante, o iodeto deve ser oxidado para um estado de oxidação superior. Isto
é dependente de peróxido de hidrogénio e catalisado pela TPO-> Enzima com heme ligado à
membrana que usa o H2O2 como oxidante. Também incorpora o Iodo na tirosina da
tiroglobulina (iodação aromática) e junta os iodotirosis da DIT para formar T4 e T3.
TSH: estimula geração de H2O2 e, por conseguinte a iodação logo aumenta a actividade da TPO.
Na membrana apical, na qual o iodeto, no lúmen do folículo é oxidado pela TPO na presença
de H2O2 para formar uma espécie activa de iodo que vai iodar tirosis da tiroglobulina.
O H2O2 é limitante da oxidação de I, da iodação aromática dos tirosis e da reacção de ligação. O
sistema gerador de H2O2 está na membrana apical e envolve a oxidação de NADPH pela
NADPH/NAD oxidase.
3) União de resíduos de iodotirosina
Catalisada pela TPO na membrana apical, e os passos ocorrem simultaneamente.
Ocorre na tiroglobulina e envolve a união de 2 anéis da DIT para formar a T4. A formação da T3
envolve um anel da MIT e outro da DIT
A localização dos resíduos iodotirosil na tiroglobulina cria um alinhamento espacial óptimo
de forma a facilitar a união
4) Proteólise da tiroglobulina e libertação das iodotironinas
A libertação das hormonas da tiroglobulina inicia-se na reabsorção da tiroglobulina por

Química Farmacêutica – PFFH – UP7
23
endocitose para a célula epitelial folicular e a proteólise completa pelas enzimas lisossomais
digestivas das células foliculares. Esta origina MIT, DIT (não saem da tiróide e são
selectivamente deiodinadas a tirosina e recicladas a tiroglobulinas e I-), T3 e T4 (secretadas na
circulação).
5) Conversão da tiroxina a triiodotironina
A T4 é secretada em maior quantidade e é
considerada como pró-hormona
Possui maior meia-vida e atinge [] mais elevadas na
circulação do que T3.
Conversão enzimática da T4 a T3: Obrigatório na
acção nos tecidos extratiroideus
Nos tecidos periféricos, 33% da T4 sofre 5’-
deiodinaçao a T3 e 40% sofre deiodinaçao do anel
interno a rT3 (inactivo)
A deiodinaçao da T4 é um processo redutivo catalisado por iodotironina deiodinases (D)
Transporte das hormonas da tiróide na corrente sanguínea
Possuem solubilidade limitada -> 3 transportadores:
TBG (thyroid binding globulin-principal), transterrina
(TTR) e albumina.
A TBG possui alta afinidade para a T4 e baixa afinidade para a T3 (importante no início rápido e
T1/2 reduzido).
Entram nas células por difusão facilitada ou transporte activo 2º devido a gradiente de Na.
T3 e T4 não se encontram distribuídas uniformemente pelas células. Grande parte de T4 é
armazenada no fígado e rins, enquanto a T3 se encontra no cérebro e nos músculos.
Metabolismo e excreção
Metabolismo periférico de T4 (fígado), ocorre por:
Desiodação do anel externo a T3, pela 5´-D.
Desiodação do anel interno a rT3, pela 5-D.
A T4 e T3 são conjugadas -> glucuronido (T4) ou sulfato (T3) com o
OH fenólico. Os conjugados de iodotreonina são excretados via biliar.
Metabolismo adicional:
Degradação da cadeia lateral seguido de transaminação, desaminação
oxidativa e descarboxilação para originar ácido tiroacético e tiroetanediol
Clivagem da ligação éter

Química Farmacêutica – PFFH – UP7
24
2. Agentes terapêuticos
2.1. Terapia de substituição - Preparações de hormonas, incluindo a origem e características
Substituição -> preparações naturais ou sintéticas contendo T4 de Na, T3 de Na ou
ambas. Grande número de compostos estimulam ou previnem a formação de hormonas pela
tiróide, por interferirem com a absorção de I nas células foliculares, inibir o TPO, prevenirem a
ligação da hormona da tiróide às proteínas ou agir como efectores da “deiodinases” da tiróide;
Divididas em 2 categorias:
Natural derivadas de tiróide animal Preparações sintéticas
Hormonas da tiróide sintéticas
As sintéticas cristalinas são mais absorvidas que as biológicas e contêm quantidades
mais precisas de princípio activo nas formas farmacêuticas.
Levotiroxina
Pela forte ligação às proteínas, o sal de Na cristalino sintético L-T4 tem início mais
lento que T3;
Leva a aumento dos níveis de T4 e, em menor grau, a aumento dos níveis de T3.
Liotironina
T3 cristalino de sódio; Rápido início de acção, mas curta duração de ação;
Quando é desejável rápido início ou cessação da actividade (ex: insuficiência cardíaca).
2.2. Fármacos antitiroideus
- Conhecer as preparações de iodo úteis como antitiroideus
Iodeto
Inibe a libertação da hormona da tiróide-> Uso no hipertiroidismo;
Com o uso dos fármacos antitiroideus, tem sido relegado para cirurgia à tiróide;
O iodeto, como solução de Lugol ou solução saturada de iodeto de K, é administrado,
durante 2 semanas, para assegurar a diminuição da vascularidade;
- Estruturas, características e REA do metimazole, do propiltiouracilo e relacionados.
Tionamidas: potentes inibidores da TPO (tiroperoxidase), responsável pela iodação da
tirosina da tiroglobulina e pela ligação de iodotirosina para formarem iodotironinas.
Mais úteis: Tioureilenos-> Derivados heterocíclicos, 5-6 membros, da tioureia, incluem:
Tiouracilo-6-n-propil-2-tiouracilo (PTU)
1-metil-2-mercaptoimidazole
A absorção pela tiróide é estimulada pela TSH e inibida pelo iodeto.
O grupo R-CS-N é designado como tioamida, tionamida, tiocarbamida ou,
se R = N, como no tiouracilo, PTU, e MMI, é designado por tioureileno.

Química Farmacêutica – PFFH – UP7
25
Esta estrutura (R-CS-N) pode existir nas formas tautoméricas tioceto ou tioenol.
REA
• Máxima atividade antitiroideia com 6-propiltiouracilo. 6-metiltiouracilo (inibe a 5’ D-I) tem
menos de 1/10 da atividade do PTU.
• Estudos REA de tiouracilos e relacionados como inibidores da deiodeinase anelar externa:
o C2 tioceto/tioenol e o N1 não substituído são essenciais para a atividade.
o OH enólico em C4 no PTU e o alquilo em C5 e C6 aumentam a potência inibitória
o Metimazole: Mais inibidor e maior duração de ação que PTU mas não é capaz de
inibir a deiodinação periférica da T4, pela presença do CH3 em N1.
o Melhorar o sabor e diminuir libertação do MMI -> 1-carbetoxi-3-
metiltioimidazole (carbimazole) -> Pró-fármaco do metimazole.
- Conhecer outras moléculas relevantes, relacionando as suas estruturas e características
físico-químicas com os seus efeitos
Bociogénicos e fármacos que afetam a função da tiróide:
• Sais de lítio: Li: Concentrado pela tiróide por transporte ativo. Iões de lítio inibem a
adenilato ciclase, que forma cAMP. Este é formado em resposta ao TSH e estimula os
processos da libertação de hormonas tiróideas.
• Compostos contendo S: Naturalmente em derivados de glucosinolatos. Quimicamente,
estes podem dar origem: Tiocianato, isotiocianato, nitrilos e tiooxazolidonas. Tiocianato ->
Anião que compete com I- para absorção pela tiróide. O seu efeito bociogénico pode ser
revertido por ingestão de I-.
• Goitrina: 5-R-viniloxazolidina-2-tiona: Potente inibidor da peroxidase tiróidea mais
efectivo que o PTU em humanos.
Outros compostos que afectam a função da tiróide incluem:
• Sulfonamidas (Ex: Carbutamida)
• Anticoagulantes (Ex: varfarina)
• Compostos aromáticos oxigenados e iodinados (Ex:Floretin[composto
oxigenado])
Exemplos de fármacos iodinados que afetam a função da tiróide são:
• Agentes antiarrítmicos(Ex: amiodarona)
• Agentes de radiocontraste (ácido ipanóico e ipodóico)
Todos estes compostos interferem com a deiodinação periférica de T4 e estão a ser testados
como adjuntos no tratamento do hipertiroidismo.

Química Farmacêutica – PFFH – UP7
26
3. Análogos das hormonas tiroideias
- REA: cadeia lateral alifática, anel com cadeia lateral da alanina; átomo “ponte”; anel
fenólico; OH fenólico.
1ºs análogos desenhados como antagonistas foram: butil-3,5-diiodo-4-
hidroxibenzoato (BHDB) -> Inibidor da deiodinase, e 2’6’-diiodotironina com
efeito antitiroideu fraco
REA
Só compostos com núcleo fenil-X-fenil substituído mostram atividade hormonal significativa.
Compostos com anéis simples (ex: DIT), derivados alifáticos e alílicos não
têm actividade como T4.
Variações estruturais na molécula T4
o Cadeia lateral alanina
o Posições 3- e 5- no anel interno
o Átomo na ponte
o Posições 3’ e5’ no anel externo
o Hidroxilo fenólico em 4’
A síntese e estudo (métodos qualitativos e quantitativos, incluindo correlação de
Hansch) de análogos às hormonas tem permitido conhecer melhor o seu mecanismo e REA.
Para existir atividade tiromimética são necessários 2 aromáticos (separados por
espaçador O, S ou C) e é necessária a disposição ortogonal (120º), imposta pela dissubstituição
por I em 3 e 5 no éter difenílico. Eliminação leva à perda da
afinidade para receptor. Excepção: análogos dibromados,
mais ativos in vivo pela maior absorção oral.
Substituição do éter por isósteros CH2 ou S->
Mantém a actividade-> Retém conformação preferida.
Bifenílos e metilenoxi: Não activos.
Triiodotironina: Substituinte em 3´pode orientar-se proximal ou distal em relação ao
resto da molécula (figura) e o OH fenólico em
4´pode ser cis ou trans em relação ao 3´.
A cadeia de tirosina pode adotar
conformações cisóides ou transóides.
OH 4´ (essencial) liga-se por ponte de
H a um centro básico aceitador de H+ do
receptor. Só pode ser substituído por NH2 (um
dos H também forma ligação intramolecular

Química Farmacêutica – PFFH – UP7
27
com I-3´). Este 4´-Oh também é importante para a ligação à proteína transportadora TBPA.
Alguns análogos sem o 4´-OH são activos-> Hidroxilados metabolicamente. Do mesmo
modo, os 4´-metoxilos podem ser pró-fármacos, activados por desmetilação metabólica.
O I em 3´pode ser substituído por muitos grupos lipofílicos (Ex: halogénio, alquilos – o
isopropilo tem efeito óptimo, arilos,...) sem diminuição da afinidade para o recetor.
A introdução de outra substituição em 5´ impede estericamente a formação da ponte
de H do OH-4´ com o receptor-> Perda de atividade (proporcional ao tamanho do substituinte).
Esta também favorece a ligação às proteínas, reduzindo a [hormona] livre.
Em 3 e 5 devem existir halogénios (I) ou metilos mantendo, os anéis em posição
perpendicular e participam em ligações hidrofóbicas com o receptor.
Cadeia lateral aniónica com 2-3 C’s, em posição para em relação ao espaçador (o anião
liga-se por ligação iónica ao recetor nuclear)-> Importante.
A amina da tirosina não é essencial para a atividade (desfavorece a afinidade para o
receptor). Os análogos sem este grupo, têm boa actividade mas baixo T1/2. Esta é importante
no transporte das hormonas e atrasa o metabolismo da hormona.
Hipotiroidismo pode ser tratado com administração regular de I mas quando a
glândula não está funcional ou destruída tem de se administrar (levo)tiroxina sintética.
Investigação direcionada para
tiromiméticos seletivos, ex: Derivado
piridazinona, tão activo como a T3 na
acção hipocolesterolémico hepática,
mas tem atividade cardíaca mínima.
Cadeia Lateral Alifática
• Hormonas naturais biossintetizadas a partir da L-tirosina possuem cadeia lateral L-
alanina.
• Os isómeros L da T4 e T3 (1 e 3 na tabela) são mais ativos que os D (2 e 4).
• O carboxilato e o nº de átomos entre ele e o anel são mais importantes para a atividade
que a cadeia lateral zwiteriónica de alanina intacta.
• Série carboxilato: Actividade máxima com cadeia lateral ácido acético de 2C (7 e 8). Diminui
quando é mais curta (ácido fórmico->5 e 6) ou mais longa (propiónico e butírico->9 a 12).
• Os com cadeia lateral etilamina (13 e 14)
são menos activos que os análogos
carboxilato.

Química Farmacêutica – PFFH – UP7
28
• Os isómeros da T3 nos quais a cadeia alanina troca de lugar com o 3-I ou ocupa a posição 2
são inactivos, indicando a localização critica da cadeia lateral na posição 1 do anel interno.
Anel com cadeia lateral de alanina
• Denominado anel interno ou anel α.
• Substituído com I em 3- e 5- na T4 e T3.
• A remoção dos I do anel interno para formar 3’,5’-T2 (15) ou 3’-T1 (16) produz compostos
sem atividade semelhante à T4-> Perda da orientação perpendicular da conformação do
difenil éter.
A retenção da actividade é observada na substituição dos I por Br (17 e 18), indicando que o I
não tem um papel único na atividade das hormonas -> A atividade encontrada em análogos
sem halogéneos (19 e 20) indica que o halogéneo não é essencial para a atividade.
• Contrariamente ao que acontece com a T3, a 3’-isopropil-3,6-dimetil-L-treonina (20)
atravessa a placenta-> -> útil em deficiências tiroideias fetais.
• Substituição em 3- e 5- por alquilos maiores e menos simétricos que os metilos (isopropilo e
butil 2º)-> Inactivos (21 e 22)->
Dissubstituição em 3 e 5 por grupos
simétricos e lipofílicos não excedendo o
tamanho do I-> Requerida para a atividade.
Átomo “ponte”
• Alguns análogos em que o O do éter foi
removido ou substituído por outros.
• O bifenilo da T4 (23) formado por remoção do O da ponte é inativo.
• A estrutura linear bifenilo está altamente alterada da conformação difenilo éter hormonal.
• Substituição do O por S (24) ou por metileno (composto 25)-> Análogos altamente ativos.
Anel Fenólico
• Denominado anel interno ou β. Necessário para atividade hormonal.
• Variações em 3’ ou 3’, 5’ têm efeitos dramáticos na actividade e afinidade para o receptor
no núcleo.
• A estrutura não substituída da série L-T2 (26) possui fraca atividade.
• Substituição em 3’ por OH polares ou nitro (27 e 28)-> Diminuição na actividade,
consequência da diminuição da lipofilia e ligações de H intramoleculares com o 4’-OH.
• Substituição por halogéneos não polares ou alquilos resulta no aumento da atividade
relacionado directamente com o impedimento e lipofilia do substituinte:
F < Cl < Br < I (29 a 31)

Química Farmacêutica – PFFH – UP7
29
CH3 < CH2CH3 < CH(CH3)2 (32 a 34)
• 3’-isopropiltironina (34) é o análogo mais potente mas o n-propiltironina (35) é muito
menos activo, pela estrutura menos compacta.
• Ao avançar na série: Actividade diminui. Maior redução no mais impedido 3’ no fenil (36).
• Substituição em ambos 3’ e 5’ pelo mesmo halogéneo-> Hormonas menos ativas (37 e 38)
que os análogos 3’-monossubstituidos (29 e 30).
• A diminuição da atividade resulta do aumento da
ionização do OH fenólico e consequente aumento
da ligação à TBG (transportador no plasma).
• 2º substituinte adjacente a OH fenólico (5’)->
Reduz actividade proporcionalmente ao tamanho.
Grupo hidroxilo fenólico
• Um OH fenólico fracamente ionizado em 4’ é essencial para atividade hormonal ótima.
• Substituição do 4’-OH por amina (39)-> Diminuição da actividade, por fracas ligações de H.
• A retenção de atividade do 4’-não substituído (40) evidencia a 4’-hidroxilação metabólica
como passo activador.
• Introdução de substituinte 4’ que não imite o papel do fenol como o metilo (41) e não é
metabolicamente convertido num resíduo funcional-> Perda completa da atividade.
• A atividade tiromimética do 4’-metil éter (42) é atribuída à rápida clivagem metabólica para
formar um análogo 4’-OH ativo.
• O pKa do 4’-OH é 6,7 para a T4 (90% ionização a pH 7,4) e 8,5 para a T3 (10%). A maior
acidez da T4 resulta da maior afinidade
para as proteínas e consequentemente
do seu > T1/2 plasmático.
- Características conformacionais das hormonas tiroideias e análogos e a sua importância
A orientação perpendicular dos planos dos aromáticos de 3,5-
diiodotironinas minimiza as interacções entre os I em 3 e 5 e os H em 2’ e
6’. Nesta orientação, 3’ e 5’ não assumem conformações equivalentes e o
I em 3’ da T3 pode ser orientado na forma distal ou proximal à cadeia
lateral que contém o anel.
Como a actividade de compostos como o 3’,5’-diiodotironina
demonstrou que alquilos podem substituir os I em 3’ e 5’-> Síntese de
compostos com alquilo em 3’ e alquilo ou I em 5’.

Química Farmacêutica – PFFH – UP7
30
A substituição em 3’ é favorável para a actividade mas a substituição em 5’ não.
Análogos distais: compostos I e II; análogos proximais: compostos III e IV
Além de ser perpendicular ao anel interno, o fenol externo pode adoptar
conformações relativas à cadeia lateral alanina, que podem ser cis ou trans.
• As conformações cisoide e transoide resultam do CH2 na
cadeia lateral alanina estar cis ou trans em relação ao fenol.
• Ambas as conformações são energeticamente similares.
D. Fármacos utilizados no tratamento de dislipidémias
1. Introdução – conhecer a estrutura do colesterol e dos triglicerídeos e compreender a
sua biossíntese, transporte e degradação no organismo humano.
Síntese e degradação do colesterol
Colesterol:
Esteróide C27; Componente importante das membranas celulares;
Precursor dos androgénios, estrogénios, progesterona, e adrenocorticóides;
Formação de pirofosfato de isopentenilo de 3 moléculas acetil CoA.
A conversão de 3-OH-3-metilglutaril-CoA (HMG-CoA) a ác.
mevalónico é importante Controlo 1º da síntese de colesterol.
Esta reacção é catalisada pela HMG-CoA redutase que reduz o
tioéster da HMG-CoA a um OH primário.
Acoplamento de seis moléculas pirofosfato de isopentenilo para
formar esqualeno.
3 moléculas de pirofosfato de isopentenilo são condensadas
para formar pirofosfato de farnesilo, um intermediário C15.
2 moléculas de pirofosfato de farnesilo são combinadas usando
uma reacção similar.
Ciclização do esqualeno a lanosterol.
Epoxidação inicial do esqualeno, seguida de ciclização que
requer um fluxo combinado de 4 pares de e- e migração de 2
metilos.
Conversão do lanosterol a colesterol.
Remoção de 3 metilos do lanosterol, redução da dupla da
cadeia lateral, movimento da outra dupla dentro da estrutura
do anel.
Requer aproximadamente 20 passos.

Química Farmacêutica – PFFH – UP7
31
O colesterol é transformado enzimaticamente por 2 vias:
Clivagem oxidativa pela desmolase (clivagem da cadeia
lateral)-> Pregnenolona, intermediário comum na síntese de
esteróides endógenos. Ou,
Conversão em ácidos biliares e sais biliares. (Mais
importante). A 7α-hidroxilase catalisa o passo inicial e
limitante desta via metabólica -> Enzima-chave de controlo.
Ácido cólico e derivados são conjugados com glicina ou taurina. Sais biliares (ex: glicocolato)
são surfactantes activos funcionando como detergentes aniónicos.
Sais biliares: Sintetizados no fígado, armazenados na vesícula biliar e libertados para o ID,
onde emulsionam lípidos da dieta e vitaminas lipossolúveis promovendo a absorção destes
compostos através da mucosa intestinal.
Reabsorvidos na circulação entero-hepática, retornam ao fígado, onde exercem feedback
negativo na 7α-hidroxilase, regulando conversões subsequentes do colesterol.
“Ácido biliar” e “sal biliar”-> Formas não-ionizada e ionizada, respectivamente. A pH
fisiológico e intestinal existem quase exclusivamente na forma ionizada.
Triglicerídeos
são formados a partir de glicerol-3-fosfato e acil CoA e acumulam-se no
citosol das células adiposas.
Quando necessários para produzir energia, são hidrolisados por lipases
para libertar AG livres que são sujeitos a β-oxidação (ciclo do ácido
cítrico) e fosforilação oxidativa.
Transporte de colesterol e triglicerídeos
Colesterol e triglicerídeos são solúveis em solventes orgânicos mas
insolúveis em fluidos aquosos, fisiológicos.
Para serem transportados no sangue, estes lípidos são solubilizados
através da associação com agregados macromoleculares – lipoproteínas.
Cada lipoproteína está associada com proteínas adicionais – apolipoproteínas (apo) – na
superfície externa.
Estas fornecem apoio estrutural e estabilidade, ligam-se aos receptores e actuam como
cofactores para enzimas envolvidas no metabolismo de lipoproteínas.
Inter-relação entre as lipoproteínas: via dividida nas componentes exógena (ingestão na dieta)
e endógena (sintética).

Química Farmacêutica – PFFH – UP7
32
Via exógena: Começa após a ingestão de gorduras. Os lípidos são absorvidos na forma de
colesterol e AG.
Os AG são reesterificados nas células da mucosa intestinal e com o colesterol são
incorporados em quilomícrons, a maior lipoproteína.
Durante a circulação, os quilomícrons são degradados pela lipoproteína lípase (requer
apolipoproteína C-II).
Os ácidos livres ficam disponíveis para armazenamento ou
produção de energia pelos tecidos adiposo e muscular.
Via endógena: começa no fígado com a formação de VLDL
(metabolismo semelhante aos quilomícrons – Lipoproteína
lipase reduz conteúdo triglicerídeo do VLDL aumentando os AG
livres no músculo e no tecido adiposo).
Lipoproteína resultante-> IDL-> Metabolizada a LDL ou
transportada para o fígado por endocitose mediada por
receptor envolvendo interacção entre o receptor de LDL com as
apolipoproteínas apoB-100 e apoE, na IDL.
A captação de LDL pelas células hepáticas e extra-hepáticas é mediada por uma interacção
do receptor com a apoB-100 na LDL.
Função do HDL (composto principalmente por apoA-I)-> Scavenger para remover o
colesterol das células extra-hepáticas e facilitar o transporte de volta para o fígado.
O HDL nascente aceita colesterol livre, não esterificado. Depois a lecitina-colesterol
aciltransferase esterifica o colesterol-> Ésteres de colesterol resultantes movem-se da
superfície para o núcleo e resulta na produção HDL3 esféricas. À medida que colesterol é
adicionado, HDL3-> HDL2, maior e menos denso.
Retorno final do HDL2 ao fígado: Transporte reverso de colesterol-> Transferência
intermediária dos ésteres de colesterol do HDL para VLDL/IDL regenerando HDL esféricas->
Recirculam e obtêm excesso de colesterol doutros tecidos.
2. Fármacos
2.1. Sequestrantes de ácidos biliares
Colestiramina- > Resina de troca aniónica, sequestra ácidos biliares no intestino e excreta-
os, aumentando a síntese no fígado por feedback.
Aumento da síntese de Ác. biliares -> Aumenta metabolismo colesterol e diminui [LDL].
Interferem com a absorção de outras gorduras e vitaminas lipossolúveis.
- Conhecer quimicamente as estruturas e compreender o seu modo de actuação.

Química Farmacêutica – PFFH – UP7
33
Colestiramina: reduz os níveis de LDL plasmáticos aumentando indirectamente a taxa a que
o LDL é depurado da corrente sanguínea.
Ligam-se ao ácido glicocólico e ácido taurocólico aumentando a excreção fecal retornam
ao fígado [] diminuídas destes compostos-> Remove a inibição por feedback da 7α-
hidrolase e aumenta a conversão hepática de colesterol a ácidos biliares.
A diminuição das [colesterol] hepáticas leva a vários efeitos compensatórios: aumento da
expressão de receptores de LDL, aumento da captação hepática de LDL plasmático, indução
da HMG-CoA redutase e aumento da biossíntese de colesterol.
Os últimos 2 efeitos são insuficientes para contrariar os aumentos na depuração e
catabolismo do colesterol e o uso concomitante de um inibidor da HMG redutase pode
fornecer um efeito adicional na diminuição do colesterol LDL.
Não alteram remoção de LDL plasmático por mecanismos não mediados por receptor.
Retorno diminuído de ácidos biliares ao fígado -> Aumento na síntese de triglicerídeos e
elevação transitória de VLDLs. Mecanismos subsequentes aumentam a remoção de VLDL,
por aumento de receptores de LDL -> VLDL volta aos níveis pré-fármaco.
- Semelhanças/diferenças estruturais e conhecer a REA dos agentes deste grupo.
Colestiramina: Copolímero constituído por
poliestireno, com pequena quantidade de
divinilbenzeno como agente de ligação cruzada.
Também contém cerca de 4 mEq de amónios
quaternários fixos por grama de resina seca.
Estes carregados positivamente funcionam como
locais de ligação para os aniões. Todos estes
locais são acessíveis aos ácidos biliares.
Aumentando a quantidade de divinilbenzeno de
2%-4% para 8%: Aumenta-se a ligação cruzada e
reduz-se a porosidade da resina-> Impede a ligação dos ácidos biliares aos locais interiores
e diminui a eficácia do composto.
- Estruturas e propriedades FQ com a FK e o potencial de efeitos adversos e de interacções.
Propriedades físico-químicas
Resinas grandes, higroscópicas, insolúveis em água.
Colestiramina: Muitas amónias quaternárias -> Muitas cargas positivas permanentes.
pKaaminas 9,0-10,15 -> Primariamente ionizados a pH intestinal.
Parâmetros farmacocinéticos, metabolismo e dosagem

Química Farmacêutica – PFFH – UP7
34
Colestiramina: Não absorvida VO nem metabolizada pelas enzimas GI. Excretada nas fezes
como um complexo insolúvel com ácidos biliares.
Disponível como pó misturado com água, sumo ou bebida não carbonatada.
2.2. Inibidores da HMGCoA redutase (HMGRIs)
O farmacóforo das estatinas assemelha-se ao substrato da HMG-CoA redutase.
Na presença destes, as células do fígado aumentam a síntese de receptores de LDL e a
captação e remoção de LDL da circulação sistémica.
- Conhecer a descoberta, saber explicar o desenvolvimento e conhecer a classificação
química das estatinas utilizadas na actualidade.
Mevastatina (compactina)-> Desenvolvimento e uso dos HMGRIs. Mevastatina
metabolito fúngico isolado de 2 espécies de Penicillium-> Inibição potente e
competitiva da HMG-CoA redutase.
Composto semelhante-> Isolado de Monascus ruber e Aspergillus terreus-> Lovastatina
(mevinolina), mais potente que a mevastatina. Difere da mevastatina pela presença de um
metilo em 6’ do anel bicíclico.
Ambas ligam fortemente à HMG-CoA redutase: Lactonas
hidrolisadas mimetizam intermediário tetraédrico da
redutase.
As porções bicíclicas ligam-se ao local CoA da enzima.
- Semelhanças/diferenças estruturais entre diferentes
estatinas, REA deste grupo e o seu mecanismo de acção.
REA
A actividade dos HMGRIs é sensível à estereoquímica da
lactona, à capacidade do anel lactona ser hidrolisado e ao comprimento da ponte que liga
os dois aneís.
Anel bicíclico-> Pode ser substituído por outros anéis lipofílicos cujo tamanho e forma são
importantes para a actividade.
Modificações no anel bicíclico e cadeia lateral éster da lovastatina-> Sinvastatina e
pravastina.
Pravastatina: ácido dihidroxi de anel aberto com 6α-OH, mais hidrofílica que a
lovastatina e a sinvastatina-> Penetração mínima nas membranas lipofílicas das células
periféricas, melhor selectividade para tecidos hepáticos e redução da incidência de
efeitos 2ºs da lovastatina e a sinvastatina.

PFFH – Química Farmacêutica – UP7
35
Substituição do bicíclico por aromáticos substituídos-> Fluvastatina,
atorvastatina, rosuvastatina.
O análogo 2,4-diclorofenilo (A) era menos potente que a mevastatina.
O pirrole substituído (B) tem menor actividade que a mevastatina e foi um intermediário-
chave no desenvolvimento da atorvastatina.
As substituições 4-fluorofenil e
isopropilo em B são observadas no
indol e pirimidina da fluvastatina e
rosuvastatina, respectivamente,
sendo substituições óptimas aí.
Os HMGRIs são ácidos 7-substituídos-
3,5-dihidroxiheptanóicos. Podem ser
subclassificados pelo anel menor.
Compostos relacionados com a
mevastatina e lovastatina têm características estruturais comuns com o anel A e os
completamente sintéticos têm características estruturais comuns com o B.
Mecanismo de acção
Diminuem os níveis plasmáticos de colesterol por três mecanismos relacionados:
1. Inibição da biossíntese de colesterol;
2. Aumento da captação mediada por receptores de LDL;
3. Redução de precursores de VLDL.
HMG-CoA redutase: Limitante na biossíntese de colesterol-> Inibição-> Diminuição inicial
do colesterol hepático.
Mecanismos compensatórios-> Aumento da expressão da HMG-CoA redutase e receptores
de LDL-> Diminuição leve a moderada da síntese de colesterol, aumento significativo da
captação de LDL mediada por receptores e diminuição global dos níveis plasmáticos de LDL.
As estatinas não diminuem os níveis de LDL nos incapazes de produzir receptores de LDL->
Aumento da expressão de receptores é o mecanismo 1º para diminuir os níveis de LDL.
O aumento dos receptores de LDL pode aumentar a remoção directa de VLDL e IDL,
precursores de LDL-> Contribui para a diminuição global do colesterol LDL plasmático.
Todos os HMGRIs podem produzir um aumento moderado de HDL.
- Propriedades FQ com a sua FK e com o seu mecanismo de acção.
Propriedades físico-químicas

PFFH – Química Farmacêutica – UP7
36
Todos os HMGRIs activos possuem um ácido carboxílico. Este é necessário para a actividade
inibitória, possui pKa de 2,5-3,5 e está primariamente ionizado a pH fisiológico.
Lovastatina e sinvastatina são pró-fármacos lactona neutros – não electrólitos.
Pravastatina, fluvastatina e atorvastatina são fármacos acídicos. Os N do indol e pirrol da
fluvastatina e atorvastatina, respectivamente, são N aromáticos não ionizáveis.O par de e-
não emparelhados-> Manutenção da aromaticidade do anel e não está disponível para se
ligar a protões.
Rosuvastatina é um composto anfotérico mas a pirimidina é fracamente básica e não se
encontra ionizada a pH fisiológico.
Atorvastatina, Fluvastatina, lovastatina
e sinvastatina: Lipossolubilidade maior
que pravastatina e rosuvastatina. Hidrólise do anel lactona dos pró-fármacos produz um
3,5-dihidroxicarboxilato que aumenta significativamente a solubilidade aquosa.
A HMG-CoA redutase é estereosselectiva. A estereoquímica 3R,5R das formas activas da
mevastatina e lovastatina é necessária para a inibição estando nos outros HMGRIs. A
estereoquímica dos substituintes nos anéis bicíclicos da lovastatina, sinvastatina e
pravastatina é menos crucial para a actividade.
Metabolismo
A maioria dos HMGRIs (incluindo formas activos dos pró-fármacos) sofrem extenso
metabolismo de 1ª passagem.
Isozima CYP3A4 -> metabolismo oxidativo da atorvastatina, lovastatina e sinvastatina.
Atorvastatina: os metabolitos orto e para-hidroxilados são equiactivos com o composto
original e contribuem significativamente para a actividade global do fármaco.
Rosuvastatina é metabolizada (limitado) pela CYP2C9 -> N-desmetilo pode contribuir para a
actividade (menos potente).
A actividade da lovastatina e sinvastatina reside no produto inicial de hidrólise (oxidação
adicional diminui a actividade).
Fluvastatina é metabolizada pela CYP2C9 e CYP3A4 a metabolitos hidroxilados activos;
estes não circulam sistemicamente e não contribuem para a actividade.
Pravastatina-> Metabolismo oxidativo-> Retêm pouca actividade, não significativos.
Parâmetros farmacocinéticos
Início de acção, duração de acção, intervalo de dosagem e ligação a proteínas semelhantes.
Atorvastatina e rosuvastatina são únicos: Duração de acção mais longa que os outros.
Ligam-se extensamente às proteínas plasmáticas. Excepção: Pravastatina (+ hidrofilica)

PFFH – Química Farmacêutica – UP7
37
Metabolismo de 1ªpassagem: BD baixa, não reflecte a absorção real.
Administração com alimentos não afecta os efeitos (Excepção: Lovastatina).
Lovastatina deve ser administrada com alimentos para maximizar a BD.
Devem ser administrados à noite-> Contrariar o pico de síntese de colesterol da manhã.
Excepções: atorvastatina e rosuvastatina Longas semi-vidas.
Eliminação: Fezes.
Extensa transformação hepática e capacidade de elevar as enzimas hepáticas: Contra-
indicados em doente hepáticos-> Elevações inexplicáveis das [aminotransferase]séricas.
- Conhecer e compreender a síntese química da sinvastatina.
Sinvastatina: Preparada de um produto de fermentação, lovastatina.
Lovastatina: Não pode ser logo convertida em sinvastatina por alquilação.
Devido à maior acidez dos α-H da lactona comparados com os α-H da cadeia
lateral éster, a alquilação ocorre preferencialmente na posição α da lactona.
2 vias conhecidas para introduzir o α-metilo adicional à cadeia 8-acilo da lovastatina.
1. Desacilação/reacilação: Desesterificação da cadeia lateral 2-metilbutanoiloxi
da lovastatina e re-esterificação com ácido 2,2-dialquilbutírico.
2. Protecção e alquilação da cadeia lateral metilbutanoiloxi com haleto de
metilo/alquilamida metálica e desprotecção.
Têm desvantagens: Nº excessivo de passos, incluindo a abertura da lactona da lovastatina,
a inserção e remoção de protectores, necessidade de
lactonização e reagentes caros.
Metilação da cadeia (S)-2-metilbutanoiloxi da
lovastatina para evitar a problemática da
desacilação/reacilação.
Protecção do C=O da lactona como um ortoformato
de forma a evitar alquilação em α da lactona.
1. Conversão da lovastatina ao seu derivado benzoato (3a) com PhCOCl e piridina em
CH2Cl2 num rendimento de 95%.
2. (3a) tratado com etano-1,2-diol, (EtO)3CH, e quantidade catalítica de H2SO4 em THF->
ortoformato (4a) desejado num rendimento de 51%.
3. Metilação da cadeia lateral (S)-2-metilbutanoiloxi de (4a) é realizada com BuLi,
pirrolidina e MeI em THF, seguida de tratamento em meio aquoso para dar o derivado
β-hidroxiortoformato (5) de sinvastatina num rendimento de 85%.

PFFH – Química Farmacêutica – UP7
38
a. Remoção simultânea do protector do OH em (4a) durante o tratamento em
meio aquoso devido à geração do meio básico em cada caso.
4. Remoção do grupo protector de C=O do composto (5) por tratamento com HCl diluído
em THF obtenção da sinvastatina num rendimento de 92%.
Principais características desta síntese: uso da protecção do C=O da lactona como os
derivados orto éster (4a) e (4b); a lactona não é aberta.
Também se obtém sinvastatina pela protecção do OH da lovastatina com cloreto de pivaloil
de forma semelhante à protecção do benzoato.
2.3. Ezetimibe – Estrutura, desenvolvimento, propriedades FQ e FK
Desenvolvimento de novos inibidores da ACAT (acil-CoA: colesterol
aciltransferase): Alvo da hipercolesterolemia.
A capacidade de C (Azetidinona) diminuir o colesterol plasmático
excede a capacidade para inibir a ACAT.
REA adicional-> Composto D e confirmação de que a actividade de diminuição do
colesterol desta classe é independente da inibição da ACAT.
Partindo de D e de dados in vivo que transformações metabólicas produziam o
composto activo que diminuía o colesterol-> Modificações-> Ezetimibe.
Alterações mais importantes:
Introdução de OHs para ajudar a localizar o composto no intestino;
Introdução de p-flúor para bloquear metabolismo indesejável.
Propriedades físico-químicas
Pó cristalino insolúvel em água mas solúvel em etanol e outros solventes orgânicos.
Fenol presente classifica-o como acídico mas este tem pKa 9,72 e está não ionizado a
pH fisiológico.
Metabolismo
Após administração oral, é rápida e extensamente metabolizado na parede intestinal e no
figado ao metabolito activo, um glucurónido fenol correspondente. Este glucurónido é re-
excretado na bílis de volta ao sítio activo.
Uma pequena quantidade sofre oxidação para dissimular o OH benzílico numa cetona.
Parâmetros farmacocinéticos
Via oral: BD absoluta não pode ser determinada pela insolubilidade aquosa e falta de
formulação injectável. Baseado na AUC, a absorção oral é moderada.
Extensa ligação plasmática do ezetimibe e do seu conjugado glucurónido.
Longa semi-vida. Coadministração de alimentos não tem afecta a extensão absorção.

PFFH – Química Farmacêutica – UP7
39
2.4. Fibratos
Bloqueiam síntese de colesterol na formação do mevalonato (Quando este exerce inibição
por feedback).
Bloqueiam também a acetil-CoA carboxilase, a enzima que produz malonil-CoA.
- Descoberta, estrutura e activação do clofibrato, os problemas e desenvolvimento de outros
fibratos com maior eficácia e segurança.
Um teste de triagem aleatório numa série de ácidos ariloxiisobutíricos demonstrou que
estes diminuem tanto o colesterol plasmático como os lipídos totais.
Melhor equilíbrio entre actividade e toxicidade: Clofibrato (etil p-clorofenoxiisobutirato),
pró-fármaco do ácido clofíbrico (ácido p-clorofenoxiisobutírico). Diminui o colesterol mas
não reduz os eventos cardiovasculares e aumenta a mortalidade global.
Clofibrato-> Protótipo para fibratos mais eficazes e
seguros.
Modificações estruturais, focadas em substituições no
anel e na adição de grupos espaçadores gemfibrozil,
fenofibrato, ciprofibrato, bezafibrato.
Mecanismo de acção
Diminuem os níveis plasmáticos de triglicerídeos mais
acentuadamente que os níveis plasmáticos de colesterol.
Diminuem muito os níveis de VLDL, causam aumento moderado de HDL e têm efeitos
variáveis nas concentrações de LDL.
Podem produzir variedade de efeitos benéficos no metabolismo de lipoproteínas:
Mediados por activação de receptores activados por proliferador de
peroxissoma (PPARs) e alteração da expressão genética.
Especificamente, os fibratos ligam-se ao PPARα.
Diminuição do VLDL plasmático:
Estimulação da actividade da lipoproteína lipase, (diminuição dos triglicerídeos
do VLDL plasmático).
Estimulação mediada por PPARα da oxidação dos ácidos gordos, inibição da
síntese de triglicerídeos e reduzida expressão de apoC-III.
Este último aumenta a acção da lipoproteína lipase (apoC-III actua como
inibidor desta enzima).
Transcrição aumentada de apoA-I e apoA-II:
Efeitos favoráveis nos níveis de HDL.

PFFH – Química Farmacêutica – UP7
40
Diminui actividade proteína de transferência de colesterol esterificado (CETP).
Todos os fibratos aceleram o turnover e remoção de colesterol do fígado: Aumenta
secreção biliar de colesterol e excreção fecal.
- Conhecer a estrutura-base dos compostos deste grupo e a sua REA.
Fibratos: Análogos do ácido fenoxiisobutírico.
O ácido isobutírico é essencial para a actividade.
Fenofibrato: contém éster-> Pró-fármaco que requer hidrólise.
Substituição em para do aromático com Cl ou isopropilo contendo Cl-: Compostos com T1/2
mais longas.
Maioria dos compostos tem ácido fenoxiisobutírico: Adição de espaçador n-propilo
(gemfibrozil): Activo.
- Conhecer as propriedades físico-químicas e a farmacocinética das moléculas deste grupo.
Contêm um ácido carboxílico de pKa 3,5-> Ionizado a ph fisiológico.
Clofibrato e fenofibrato-> Pró-fármacos éster neutros-> Não electrólitos.
Fenofibrato é lipossolúvel, pelo éster isopropilo e o aromático adicional.
Todos os fibratos
são aquirais.
Metabolismo
O fenofibrato sobre hidrólise-> Ácido fenofíbrico, activo que pode ser metabolizado por
vias oxidativa ou conjugação. Oxidação (CYP3A4) dos metilos aromáticos produz análogos
hidroximetilo e ácido carboxílico inactivos.
Fibratos e análogos oxidados são excretados como conjugados glucurónido na urina.
Parâmetros farmacocinéticos
Fibratos têm excelente BD e estão extensamente ligados às proteínas.
Os alimentos podem melhorar a absorção-> Tomados com ou antes das refeições.
2.5. Ácido nicotínico e derivados – conhecer as estruturas, o seu desenvolvimento e a sua
importância, as suas propriedades físico-químicas e a PK.
Ácido nicotínico e derivados, em [] elevadas, influenciam o rácio de
lipoproteínas, diminuindo as [] de VLDL e LDL, mas não têm efeito
nos complexos colesterol-HDL.
Ácido nicotínico (niacina) -> Sintetizado por oxidação da nicotina.
Metabolito nicotinamida -> Característica estrutural necessária do NADP+, co-factor
envolvido no transporte de e- e metabolismo intermediário.
[Ácido nicotínico] elevadas diminuem os níveis de colesterol e triglicerídeos.

PFFH – Química Farmacêutica – UP7
41
Propriedades físico-químicas
Pó cristalino branco, estável, não higroscópico.
O ácido carboxílico tem pKa de 4,76-> Ionizado a pH fisiológico.
O N da piridina é base fraca (pKa 2,0)-> Principalmente na forma não-ionizada.
Livremente solúvel em soluções alcalinas.
Metabolismo
Ácido nicotínico é uma vitamina do complexo B convertida a nicotinamida, NAD+ e NADP+
(coenzimas para reacções de oxidação/redução).
É metabolizado a vários compostos inactivos (ácido nicotinúrico, derivados N-metilados,…).
Regulação bioquímica normal e feedback evitam que grandes doses de ácido nicotínico
produzam excesso de NAD+ e NADP+.
Pequenas doses de ácido nicotínico são excretadas como metabolitos, doses maiores
excretadas inalteradas pelo rim.
Parâmetros farmacocinéticos
Prontamente absorvido. T1/2 =1h.
Monitorização: Avalia eficácia e prevenir toxicidade até se atingir dose estável e eficaz.
E. Estrogénios, progestagénios e androgénios
• Estrogénios naturais: Esteróides 18C com anel A planar insaturado com
um 3-fenólico.
• Progestagénios naturais: Esteróides 21 C e têm em comum um 3-ceto-
4-eno no anel A e uma cetona em C21. Progesterona é a mais potente
• Androgénios: Esteróides 19C e que têm em comum O em C3 e 17 (OH
ou cetona). Mais potente no sangue: Testosterona. DHT-> Metabolito mais potente em
tecidos alvo.
1. Agonistas e antagonistas de estrogénios.
1.1. Biossíntese e metabolismo – Estrutura base dos estrogénios, biossíntese e metabolismo
Estrogénios na mulher são:
1. Estradiol (o mais potente, 10-20% do estrogénio em circulação).
2. Estrona (10x menos potente que o estradiol, 60-80% do estrogénio circulante).
3. Estriol (muito fraco, 10-20% em circulação)
Estrona: 1º a ser isolada na forma cristalina a partir da urina
Biossíntese:
No folículo dominante maduro e no corpo lúteo nas mulheres pré-menopausa.
Gravidez-> Placenta, principal local síntese estrogénios.

PFFH – Química Farmacêutica – UP7
42
50% Produção de estrona ocorre nos ovários. Restante: Produzida do estradiol: Sulfato de
estrona-> Estrona na glândula adrenal; e por aromatização da androstenediona
Antes da menopausa: Rácio estrona-estradiol= 1:2
Depois: Rácio=2:1-> Perda da função do ovário.
Do colesterol armazenado nos tecidos, quando
estimulados pela gonadotropina, forma-se
estrogénios, progesterona e androgénios.
Nos ovários: FSH atua nos folículos pré-ovulatórios
estimulando a síntese dos estrogénios. Células da
teca: Colesterol-> Androgénios (células granulosas) -
> Estrogénios.
Clivagem da cadeia lateral do colesterol->
Pregnenolona que pode ser convertida a
progesterona, ou em diversos passos, aromatizar o
anel A que se encontra nos estrogénios.
Prenenolona (17α-hidroxilase)-> 17 α-hidroxipregnenolona (b), convertida ao intermediário
desidroepiandrosterona (DHEA) pela 17, 20 –liase.
DHEA-> Androstenediona (17-cetoesteróide- c e d) pela 5-eno-3β-hidroxiesteróide
desidrogenase e 3-oxoesteróide-4,5-isomerase. DHEA é interconvertível com a testosterona
pela 17β-hidroxiesteróide desidrogenase (f)
Por fim: Androgénios com 19C-> Estrogénios com 18C, por perda do CH3 angular C19 e
aromatização do anel A (aromatase)-> 17β-estradiol ou estrona (g).
Interconversão do 17β-estradiol-> Estrona pela estradiol desidrogenase (h)
(uma 17β-hidroxiesteróide desidrogenase).
Metabolismo:
Conjugados glucoronido e sulfato hidrossolúveis, no fígado. O reservatório
de estrogénios deve-se á reciclagem entero-hepática (dessulfatação e ressulfatação).
17β-estradiol exógeno sofre 50% de metabolismo de 1º passagem a metabolitos excretados
na urina. Restante: No fluido biliar-> 10% encontrada nas fezes,
restante retorna ao fígado.
17β-estradiol e Estrona: Convertidos do complexo enzimático
CYP450 16α-hidroxilase em estriol encontrado na urina como
conjugado glucorónido.

PFFH – Química Farmacêutica – UP7
43
Estrogénios: Metabolizados por hidroxilação em orto em relação ao 3-fenolico, pela 2/4-
hidroxilase dos estrogénios (CYP3A4), para dar 2-hidroxiestrogénios e 4-hidroxiestrogénios.
Estes metabolitos ligam ao receptor de estrogénios mas têm actividade fraca/moderada.
São instáveis: Convertidos ao 2-metoxi e 4-metoxi estrogénios e a conjugados glucoronido
encontrados na urina.
37% Estradiol circulante liga-se á globulina de ligação a hormonas sexuais (SHBG) e 61%
liga-se á albumina. 2% Livre. Estrona e Ésteres
de sulfato da estrona e do estradiol possuem
menor afinidade para a SHBG e maior para a
albumina.
São essenciais para o transporte, distribuição e clearance dos estrogénios.
Estradiol é capaz de induzir a síntese de SHBG
1.2. Agonistas de estrogénios
1.2.1. Esteróides – Estruturas dos compostos, potência e farmacocinética
Estrogénios esteróides
Estradiol:
Mais potente endógeno; Alta afinidade para o receptor de estrogénios (ER);
Oralmente é rapidamente conjugado no intestino e oxidado no fígado: Baixa BD e eficácia
Etinil estradiol e mestranol
Impedimento da oxidação metabólica do OH em C17 do estradiol a estrona aumenta a BD
oral do estradiol-> Alquilação da posição C17 (ex: etinil estradiol, EE) com um alcino inerte-
> Análogo mais potente que o estradiol.
Rápida e quase completamente absorvido, com BD oral=40% e t1/2= 26h.
Extenso metabolismo de 1ª passagem a 3-O-glucuronidos e 3-O-sulfatos e hidroxilação
aromática a 2-hidroxietinilestradiol e ao O-metilado.
Ciclo entero-hepático extensivo. Bactérias do TGI
hidrolisam os glucuronidos e sulfatos, ocorrendo
reabsorção. AB-> Diminuem eficácia contraceptivos orais.
Mestranol: Éster 3-O-metil do EE, rapidamente
convertido após administração oral.
Ésteres de estradiol:
Para longa duração-> 17β-Estradiol convertido em pró-fármaco éster, administrado via IM.
A hidrólise lenta liberta estradiol durante longo período de tempo:
17β- valerato-> 14-21 dias.

PFFH – Química Farmacêutica – UP7
44
17β-ciclopentilpropionato (cipionato)-> 14-28 dias.
Estrogénios conjugados:
Grávidas-> 2 estrogénios únicos: Equilenina e Equilina, excretados na urina como conjugados
sulfato de sódio. Estes foram utilizados em preparações de estrogénios.
1.2.2. Não-esteróides - derivados do estilbeno
- Estruturas, características, semelhanças/diferenças em relação aos estrogénios naturais
Núcleo esteróide: Não é requerido para acção dos estrogénios -> Derivados do estilbeno
(difeniletileno) têm actividade estrogénica.
Dietilestilbesterol (DES): trans 10x mais potente que o cis -> Assemelha-se mais ao estradiol
O trans-dietilestilbestrol (DES) e derivado reduzido hexestrol têm semelhanças ao
esqueleto esteróide. Os 2 etil não são indispensáveis para a actividade estrogénica -> 4
metils conferem actividade comparável.
A “densidade” da molécula é importante para a sua actividade:
1) Os dois fenil planos devem ter um ângulo de torção de 60º.
2) 4 alquil fornecem mesmo impedimento que os dois etil -> Derivado dimetil é inactivo.
Distância interatómica entre os 2 OH fenólicos
do DES: Importante-> Relacionada com a
distância entre 3-OH e 17-OH do estradiol.
É 12,1 Å no DES e 10,9 Å no estradiol.
Diferença pode ser eliminada por ligações de
H de 2 H2O ao 17-OH do estradiol e por
considerar mais o OH da água em vez do OH
do estradiol.
- Conhecer a síntese do dietilestilbestrol.
Alquilação da desoxianisoína com iodeto de etilo, em base forte.
Produto + Brometo de
etilmagnésio-> Olefina trans e
pequena quantidade de cis.
Desproteção dos ésteres metílicos->
Dietilestilbesterol cuja
hidrogenação catalítica é
estereoespecifica dando o isómero
(±)-treo do hexestrol, activo.

PFFH – Química Farmacêutica – UP7
45
1.2.3. REA e farmacocinética dos estrogénios –principais aspectos
Aromático A e o OH em C3 do núcleo esteróide-> Essenciais para a actividade.
OH: Estabelecimento de uma ponte H.
Distância entre os OH dos C3 e C17 e a presença de uma estrutura planar são importantes
na estrutura e optimizam a actividade. Essa distância deve ser entre 10.3 e 12.2 Å.
Substituintes no núcleo estrogénico-> Modificam a actividade:
Anel A, qualquer função em C1-> Reduz muito a actividade.
Apenas grupos pequenos podem ser colocados em 2 e 4.
Introdução de OH em 6,7 e 11-> Diminui a actividade.
Remoção dos grupos com O em 3 ou 17 ou a Epimerização do 17β-OH do estradiol para a
configuração α-> Composto menos activo
Estrogénios equina-> Insaturações adicionais no anel B (uma a mais na equilina e 2 no da
equilinina). Esta insaturação B aumenta a potência.
Substituintes em 11β-> Bem tolerados. Ex: 11β-metoxi ou 11β-etil possuem maior afinidade
para ER que o estradiol.
Modificações em 17α e 16-> Podem melhorar a actividade:
17α-etinil ou 17α-vinil: Aumentam actividade.
Presença de grupos mais polares-> Pouco tolerada.
Em 16 são tolerados grupos com tamanhos e polaridade moderada
Aumento do anel D (Ex: D-homoestradiol)-> Diminui muito a actividade estrogénica.
Farmacocinética:
BD varia em função da via de administração:
Subcutânea: Ordem de BD
é: estradiol>estona>estriol
Oralmente:
estriol>estradiol>estrona.
Todos têm baixa BD oral ->
Análogos.
EE: Efectivo VO pela resistência
ao metabolismo no TGI e fígado.
Outros estrogénios activos
oralmente: Análogos com éter
lábil (3-(2-tetrahidropiranil) e
17β-(2-tetrahidropiranil)

PFFH – Química Farmacêutica – UP7
46
estradiol, respetivamente 12 e 15x mais activos que o estradiol.
1.3. Antagonistas de estrogénios – Estruturas, características e modo de actuação:
Há 3 classes:
Interferem com a activação do receptor (estrogénios impedidos e análogos do
trifeniletileno)
Limitam a biossíntese (inibidores da aromatase)
Estrogénios “impedidos” (ex: Estriol)
Interagem com ES nos tecidos e dissociam-se rapidamente-> Impossibilidade de produzir
efeito estrogénico.
Em elevadas []-> Competem ou impedem acesso do estradiol diminuindo o seu efeito.
Análogos do trifeniletileno: estruturas e características
Desenvolvimento de moduladores selectivos do receptor dos estrogénios (SERM) que
exibem actividade agonista ou antagonista específica para um tecido.
3,4-dihidronaftaleno e benziotiofeno nos SERMs-> Análogos rígidos de trifeniletilenos.
1º SERM utilizado: Benzotiofeno raloxifeno, agonista usado na
osteoporose.
Antiestrogénios trifeniletileno: Estruturalmente idênticos á família
estilbeno dos estrogénios tendo elevada afinidade para o ER.
Impedem a translocação do complexo do ER para o interior do
núcleo das células alvo e interferem com a ligação do complexo
hormona-receptor ao local aceitador na cromatina
Mecanismo molecular de ação dos SERMs:
ER deve ser alvo primário.
Quanto mais células possuírem ER melhor o tumor responde á terapia antiestrogénica.
ER: Dividido em 6 regiões (A-F). A região de ligação ao DNA (C) é essencial para a interação
com elementos de resposta aos estrogénios.
Domínio de ligação ao estrogénio (E)-> Onde os inibidores competitivos se ligam
Regiões de função de activação (AF-1 e AF-2): Interagem com moléculas co-activadoras
para formar unidades efectivas de transcrição em genes de reposta a estrogénios.
Cristalização do domínio de ligação (E) do ER quando ligado ao estradiol, DES, raloxifeno e
4-hidroxitamoxifeno-> Considerações sobre mudanças conformacionais aquando da ligação
de agonistas e antagonistas
Diferença mais importante: Posição da hélice 12-> Essencial para a ligação dos co-
ativadores para formar o complexo de transcrição no gene de resposta a estrogénios.

PFFH – Química Farmacêutica – UP7
47
Estradiol faz com que a hélice 12 fixe o ligando dentro da bolsa hidrofóbica do domínio de
ligação e esta conformação permite a ligação do co-ativador
Raloxifeno e 4-hidroxitamoxifeno impedem que a hélice 12 sele a bolsa
hidrofóbica-> Transcrição de genes impedida-> Co-ativadores não podem ligar.
Acções antiestrogénicas e algumas semelhantes aos estrogénios dos SERMs->
Explicadas por diferentes conformações dos complexos α do ER que atrai novos
co-ativadores ou co-repressores para modularem a acção dos estrogénios nos
diferentes locais. Os recetores seriam os mesmos nos diferentes locais sendo
os co-ativadores e os co-repressores distribuídos de forma diferentes nos
diferentes locais.
ER-α pode ser modulado por diferentes [] de ER-β em locais diferentes-> ER-β
pode aumentar a activação de genes tipo estrogénio por interacções proteína-
proteína. Não é claro como a acção SERM é modulado em cada alvo: Pode
ocorrer mais de 1 mecanismo
Citrato de Clomifeno
VO como mistura de 2 isómeros geométricos, o Z (cis, zuclomifeno) com actividade
estrogénica e o E (trans, enclomifeno) que exibe actividade antiestrogénica
Tamoxifeno (SERM)
Fraca actividade estrogénica em tecidos como o endométrio e o osso.
N-desmetilação pela CYP3A4 ao metabolito principal, N-desmetiltamoxifeno e CYP2D6 ao 4-
hidroxitamoxifeno (activo) e com maior afinidade de ligação para o ER que o tamoxifeno.
A T1/2 do metabolito desmetilado (14 dias) é 2x a do tamoxifeno
O éter aminoetil na cadeia lateral é essencial para a actividade
antiestrogénica.
BD oral=100%, sofrendo metabolismo de 1º passagem mínimo e elevada
ligação á albumina.
O t1/2 elevado deve-se á ciclo enterohepático, elevada ligação proteica
(cuidados na terapia anti-coagulante) e auto-inibição metabólica
Toremifeno
Relacionado com o tamoxifeno, diferindo apenas na cloração da cadeia lateral etil que
leva a redução da potência antiestrogénica.
BD oral=100%, com mínimo metabolismo de 1º passagem e ligado á albumina. N-
desmetilação pelo CYP3A4 para formar o metabolito activo: N-desmetiltoremifeno e
sofre desaminação e hidroxilação para formar ospemifeno.

PFFH – Química Farmacêutica – UP7
48
T1/2=5-6 dias e uma vez que são
metabolizados no fígado->
Precaução em doente hepáticos
- Conhecer a síntese química do
tamoxifeno.
Semelhante á do dietilestilbesterol
- Inibidores da aromatase: Mecanismo da aromatase e a inibição por derivados esteróides e
não esteróides.
Derivados androstenediona
Os inibidores da aromatase bloqueiam a conversão dos
androgénios a estrogénios;
Competem com a androstenediona para o sítio activo da
aromatase e segundo a REA: melhores agentes são
análogos dos substratos com pequenas modificações no
anel A e C19
Análogos com aril em 7α têm afinidade aumentada para a enzima
Os seguintes têm actividade inibidora irreversível in vitro:
4-hidroxiandrostenediona
Várias androsta-1,4-dieno-3,17-dionas
10β-propinilestr-4-eno-3,17-diona
Exemastane
Inibidor irreversível baseado em esteróides com estrutura semelhante á androstenediona
Liga-se irreversivelmente á aromatase e é classificado como um inibidor suicida e possui
elevada afinidade e alta seletividade para a aromatase
VO: 50% da dose é absorvida-> Aumentada se ingerida após refeição rica em gorduras
Ligação proteica de 90% e sofre extenso metabolismo pela CYP3A4
Derivados triazole
Baseados na aminoglutenemida (inibidor da aromatase) incluem o
anastrazole e o letrozole
Inibição resulta do N4 do triazole que interage com o Fe do heme do CYP 19 (Aromatase)
Anastrazole e Letrozole: Inibidores competitivos cuja seletividade inibe a conversão da
testosterona a estrogénios reduzindo as []de estrona, estradiol e sulfato de estrona.
Diminuição da []de estrogénios-> Evita progresso de cancros dependentes de estrogénios.
Letrazole – Não esteróide.

PFFH – Química Farmacêutica – UP7
49
Inibição da aromatase é competitiva e altamente específica sem efeitos na acção das
enzimas responsáveis pela produção de glucocorticóides e mineralocorticóides
Rapidamente absorvido no TGI, não afectado pela comida.
Metabolizado pelo CYP3A4 eCYP2D6 a agente inactivo, glucorinado e excretado na urina
Inibe fortemente CYP2D6 e moderadamente CYP2C19. Tamoxifeno induz metabolismo.
Anastrozole
Derivado benziltriazole compete com o substrato para o sítio activo, sendo reversível
Reduz níveis de estradiol sem afectar os de cortisol ou aldosterona
Bem absorvido VO e eliminado na bílis. T1/2 de 50h
60% da dose oral é metabolizado no fígado por N-desalquilação, hidroxilação e
glucoronidação a metabolitos triazol inactivos
Outros
Antiestrogénios puros
Resistência à terapia com tamoxifeno deriva da actividade agonista no ER e consegue inibir
a activação da via AF-2 mas não consegue a via AF-1
Não podem ser classificados com SERMs (sem actividade agonista).
Fulvestrant
Análogo do estradiol com cadeia lateral hidrofóbica em 7α
Fraca BDoral pela fraca solubilidade aquosa
Metabolizado a derivados activos e inactivos como os esteróides endógenos
Acolbifene
Antiestrogénio não esteróide. Pró-fármaco oral, hidrolisado ao análogo
trifeniletileno rígido (activo). 250x mais potente que o tamoxifeno
Assemelha-se ao raloxifeno com um benzopireno a substituir o benzotiofeno
2. Agonistas e antagonistas de progestagénios
2.1. Biossíntese e metabolismo – conhecer a estrutura química base dos progestagénios
humanos e a sua biossíntese e metabolismo
Biossíntese
Biossíntese de progesterona: Iniciada quando a LH (secretada da hipófise anterior), se liga
ao receptor de LH da célula alvo. A activação deste receptor resulta em:
Aumento intracelular de cAMP por ativação da proteína G e adenilciclase;
Activação da esterase de colesterol, por aumento da [cAMP] intracelular, catalisa a
clivagem de ésteres de colesterol-> Colesterol livre-> Pregnolona na mitocôndria

PFFH – Química Farmacêutica – UP7
50
Pregnolona-> Progesterona pela 5-eno-3β-hidroxiesteroide desidrogenase e 3-
oxoesteróide-4,5-isomerase (passos c,d na fig 46.3)
Metabolismo
Progesterona: Metabolizada pelo fígado, tendo curto
T1/2 (5-10min).
Excretado via renal, como conjugado glucoronido ou
sulfato do 5β-pregnanediol.
Glucoronido: Índice da actividade do corpo lúteo, e
baixa prematura dos níveis -> Possível aborto
espontâneo.
Outras vias: Redução da 20-cetona e 6α-hidroxilação,
cujos metabolitos não exibem atividade progestogénica significante.
Formação do 5β-pregnanediol da progesterona: Caracterizada pela redução da dupla em 4-
5, para criar junção cis dos anéis A/B, e por redução da 3-cetona a um 3-α-OH.
No soro, a progesterona está ligada à globulina de ligação ao cortisol e à albumina.
2.2. Agonistas de progestagénios
- Classificação estrutural dos progestagénios, progestagénios mais utilizados na prática
clínica e características mais relevantes para a sua farmacocinética e actividade biológica.
Mecanismo de acção-> Interacções moleculares
Receptor: Ligando que age como activador da transcrição e faz parte da superfamília dos
receptores nucleares que possui 3 domínios conservativos funcionais:
1. domínio de ligação ao DNA
2. domínio N-terminal
3. domínio C-terminal de ligação ao
ligando.
Domínios de ligação ao DNA: 2 Zn fingers assimétricos com 4 cisteínas coordenadas com
cada Zn
Entre estes “dedos” existe uma hélice α específica para as interacções com o DNA
Domínio de ligação ao ligando: 12 hélices α e 4 folhas β que formam uma cavidade em que
o agonista se liga.
Progesterona se liga-> Alteração conformacional-> Transforma num factor de transcrição
Receptor sofre dimerização para dar PR-A e PR-B-> Actividade e estrutura diferentes.
Dímero interage com sequências específicas do DNA dentro de genes de resposta a
progesterona. Combina-se a co-ativadores que se ligam ao complexo para a transcrição de
genes cuja velocidade tanto pode aumentar como diminuir
REA

PFFH – Química Farmacêutica – UP7
51
Actividade restrita a moléculas com o núcleo esteróide
Os progestagénios sintéticos podem dividir-se em 2 classes de compostos:
Androgénios (derivados 19-norandrostano ou estrano)
17-hidroxiprogesteronas
Na série dos androgénios, Grupo 17α como: Etinil, metil, etil e
variações-> Acção oral
Etisterona (17β-hidroxi-17α-etinilprogesterona) por via
subcutânea tem 1/3 da atividade da progesterona mas tem 15x
mais actividade que a progesterona VO. Pela similaridade com a
testosterona, este progestagénio tem atividade androgénica. A
remoção do CH3 em 19-> Noretisterona (noretrindona), 5-10 x
mais potente como progestina.
Actividade da noretisterona é aumentada pela adição de Cl em 21 (bloqueiam a
hidroxilação metabólica) e adição de metilos em C18 (norgesterol)
Acetato de etinodiol: Progestagénio oral potente, mais ativo quando VO que parenteral.
Insaturações nos anéis B e C dos 19-androstanos-> Aumentam a actividade como
progestagénios.
Introdução halogénios ou CH3 em 6α e 7α-> Aumenta actividade hormonal
Acetilação do 17β-OH da noretisterona-> Aumenta duração.
Remoção do 3-ceto-> Remove efeitos androgénicos e retém forte actividade progestagénia.
Actividade das 17α-hidroxiprogesteronas é aumentada pela insaturação dos C6 e C7 e CH3
ou halogénio em C6 e pela introdução de CH3 em C11. Estes impedem a redução
metabólica dos 2 carbonilos e a oxidação metabólica no C6.
Substituintes F no C21-> Impedem hidroxilação metabólica e aumentam actividade oral.
Progesterona e análogos
Limitações na administração da progesterona pela baixa BDoral.
Para ter actividade terapêutica-> Dada por injeção ou por via intravaginal, como tal
foi necessário o desenvolvimento de derivados análogos oralmente ativos.
Progestagénios sintéticos
Problema dos 1ºs Progestagénios: Tinham efeitos androgénicos. O
desenvolvimento focou-se no aumento da actividade para o Receptor da
Progesterona, com mínimo de actividade androgénica, estrogénica ou
glucocorticoide. Contêm núcleo pregnano ou andostrano.
Progestagénios podem ser classificados em termos de gerações:

PFFH – Química Farmacêutica – UP7
52
1ª geração: Análogos 17-hidroxiprogesterona, noretinodrel, noretisterona e derivados.
2ª geração: Norgestrel e Levonorgestrel.
3ª geração: Derivados do levonorgestrel (desogestrel, gestodeno, e norgestimato).
4ª geração: 19-norprogesteronas (acetato de nomegestrol e trimegestona).
Noretisterona (noretrindona) – 19 noresteróide, um dos primeiros usados.
Combinada com estrogénios, é eficaz como contraceptivo oral. Fraco agonista androgénio,
sem actividade glucorticóide e antimineralocorticóide.
Acetato de noretisterona VO-> Completa e rapidamente
desacetilado por metabolismo de 1º passagem intestinal e
hepático a noretisterona, com BDoral=64%.
Passos seguintes: Redução da dupla em 4-> 5α e 5β-
dihidronoretisterona, bem como a redução da cetona.
36% da noretisterona liga-se á SHBG, 61% á albumina
Acetato pode ser administrado por via transdérmica com estradiol, como ciclo contínuo de
regime de substituição hormonal.
Norgestrel e levonorgestrel
Norgestrel: Formulado como mistura racémica, apenas o levonorgestrel seja activo.
Tem alguma actividade androgénica, e nenhuma glucocorticóide e antimineralocorticóide.
Pode ser administrado oralmente, via transdérmica (com estradiol em adesivo de 7 dias) e
em usos contínuos com DIU. A BDoral é de 95%.
48% ligada à SHBG, 50% à albumina. Sofre redução da cetona e hidroxilação.
Desogestrel
Pró-fármaco rapidamente metabolizado na mucosa intestinal, e na 1ª passagem no fígado,
ao metabolito activo – etonogestrel (3-cetodesogestrel).
BD oral = 84%.
Selectividade para o PR, e baixa actividade androgénica, e não diminui os efeitos benéficos
dos estrogénios nos perfis lipídicos.
Gestodeno
Não é um pró-fármaco. Ele exibe uma BD oral = 100% excelente afinidade para o PR.
Drospirenona
Derivada da espironolactona, única progestina com actividade antimineralocorticóide.
Afinidade para o receptor mineralocorticóide é 5x superior à aldosterona.

PFFH – Química Farmacêutica – UP7
53
Actividade progestogénica: 10% da verificada para o levonorgestrel. Tem actividade
antiandrogénica ao bloquear a ligação da testosterona aos AR. Não tem actividade
estrogénica nem glucocorticoide
Baseado no androstano, possui diversos grupos: dois ciclopropil, um que inclui os C6 e C7 e
outro os C15 e C16 e uma lactona em C17.
2.3. Antagonistas de progestagénios – conhecer a estrutura e principais características
Mifepristona
Compete com a progesterona para o receptor, impedindo a ligação e a
activação.
VO: Rápida absorção, com pico de [] plasmática após 90min, BDoral=70%, e T1/2=18h. 98%
encontra-se ligado às proteínas (albumina e glicoproteína ácida α1).
Metabolizada pela CYP3A4: Mono e di-N-desmetilação e hidroxilação na cadeia 17-propinil.
Eliminação biliar -> 83% Encontrado nas fezes.
Actividade antiglucocorticóide e outro análogo antiprogestogénio, como a onapristona,
exibe menor actividade antiglucocorticóide.
Estrogénios e progestagénios de acção combinada
Tibolona
19-norpregnano com acção estrogénica, progestagénica e androgénica
Pró-fármaco, metabolismo no TGI aos metabolito ativos (4insaturado, 3α-OH, e 3β-OH)
3. Agonistas e antagonistas de androgénios
3.1. Biossíntese e metabolismo – conhecer a estrutura química base dos androgénios
humanos e a sua biossíntese e metabolismo
Biossíntese
Androgénios: Sintetizadas do colesterol nos testículos e intermediários na síntese de
estrogénios. No fígado, os androgénios são formados a partir de esteróides com 21 C.
Pequenas quantidades sintetizadas no córtex adrenal, hipotálamo e adenohipófise.
• LH: Liga-se aos receptores nas células de Leydig para iniciar a síntese de testosterona e
activa a proteína Gs que aumenta o cAMP intracelular por ativação da adenilato ciclase
• O cAMP activa esterases de colesterol (libertação de colesterol livre)
• Colesterol livre-> Pregnenolona na mitocôndria por clivagem da cadeia lateral
• Esta é convertida pela 17α-hidroxilase a 17α-pregnenolona e depois a
desidroepiandrosterona (DHEA, um esteróide C-19) pela 17-20 liase, clivando a ligação do
C-17 a C-20 e perda da cadeia lateral 17β-acetil.

PFFH – Química Farmacêutica – UP7
54
• 17α-hidroxilase (na membrana do RE), composta pelo
citocromo P45017α e NADPH-citocromo P450 redutase.
• A activação da 5-eno-3β-hidroxiesteróide desidrogenase e 3-
oxosteroide-4,5-isomerase é responsável pela conversão da
DHEA ao androgénio 17-cetoesteroide, androestenediona (e
pela conversão da pregnenolona a progesterona).
• Testosterona (TRT)-> Formada por redução da 17-cetona da
androstenediona pela 17β-hidroxiesteroide desidrogenase.
• A testosterona e a androstenediona são interconvertíveis.
• A perda do CH3 angular C-19 e aromatização do anel A da
testosterona ou androstenediona é catalizada por um complexo microssomal do citocromo
P450, chamado de aromatase, e resulta respectivamente nos
esteróides C-18: 17b-estradiol e Estrona (interconvertíveis pela
desidrogenase do Estradiol).
• Androstenediona: Substracto preferido para a aromatização,
sendo usados neste processo 3 NADPH e 3 O2 para a conversão
de um androgénio a um estrogénio.
• Androgénio endógeno mais potente: dihidrotestosterona (DHT)-> Esteróide 5-alfa reduzido,
sintetizada por 2 isoformas 5-alfa redutase (tipo 1 e 2) a partir da TRT. Esta enzima é
encontrada na mitocôndria e na membrana nuclear catalisando uma redução irreversível
que precisa de NADPH e que fornece α-H ao C5.
• Tipo 1: Responsável por 1/3 da DHT em circulação. Glândulas sebáceas da pele, couro
cabeludo e fígado.
• Tipo 2: 2/3 da DHT circulante. Próstata, vesículas seminais, epidídimos, pele genital
(escroto), folículos capilares e fígado
Metabolismo da Testosterona.
1. Convertida a metabolitos activos. Na próstata, pele e fígado é reduzida a DHT pela 5α-
redutase (irreversível).
2. Pequena quantidade convertida a estradiol pela aromatase (irreversível), por clivagem do
CH3 C-19 e aromatização do anel A (Tecido adiposo ou ovários das mulheres).
• 80% Estrogénios no homem provêem da aromatização no tecido adiposo. Restantes são
secretados pelas células de Leydig nos testículos
3. Inactivada no fígado por redução e oxidação, seguida de glucoronidação e excreção renal.
4. Metabolizada a androstenediona por oxidação do 17-beta-OH e depois a androstanediona
por redução 5-alfa do anel A. Esta pode passar a androsterona, por redução do 3-ceto.

PFFH – Química Farmacêutica – UP7
55
• A androstenediona também pode ser convertida a etiocolanolona (redução 5-beta e 3-
ceto).
• DHT também pode ser convertida a androstanediona,
androsterona e androstanediol.
• 90% TRT-> Eliminada via renal, 6% recuperada nas fezes
(circulação enterohepática).
Metabolitos urinários:
• Androsterona e o diastereoisómero 5α-etiocolanolona
ambos inactivos são os principais
• Conjugados glucoronídeos, e em menor extensão como
conjugados sulfato
• A redução da TRT á conformação A/B no etiocolanolona
explica a completa perda de actividade porque anéis cis A/B não possuem afinidade para o
recetor de androgénios (AR)
• A maioria dos metabolitos sofre extensa glucuronidação no 3α e 17β-OH quer no tecido
alvo quer no fígado.
• VO: T1/2 da TRT é inferior a 30min pelo extenso
metabolismo hepático (90% sofre metabolismo de
1º passagem antes de atingir a circulação sistémica).
• Metabolitos menores na urina: 5α-androstanos e
5β-androstanos com 3α-OH
• Maiorias dos 17-cetoesteróides isolados resultam do
catabolismo dos adrenocorticóides e não dos
androgénios
3.2. Agonistas de Androgénios
3.2.1 Androgénios Esteróides
- Para os androgénios naturais e sintéticos, reconhecer as estruturas e características e
relacioná-las com a actividade androgénica e com a via de administração
Terapia de substituição de testosterona (TRT)
É difícil: falta de actividade oral de preparações com
eficácia; falta de perfil de segurança -> Estudos que
separem funções desejáveis (reguladas por androgénios
endógenos) de não desejáveis. Actualmente: preparações

PFFH – Química Farmacêutica – UP7
56
injectáveis de ésteres de testosterona e de libertação transdérmica ou bucal (na gengiva).
Ésteres de testosterona injectável (enantato de testosterona, propionato ou cipionato)
produzem flutuações indesejáveis nos níveis sanguíneos de testosterona, com concentrações
suprafisiológicas ou subfisiológicas -> benefícios satisfatórios ou efeitos indesejáveis
As preparações orais (fluoximesterona e 17α-metiltestosterona) não são usadas por
causa da toxicidade no fígado ligada ao grupo 17α-alquilo e à menor eficácia
Via Oral: rápido metabolismo de 1ª passagem -> Só pequenas quantidades atingem a
circulação sistémica. É convertida em 17-cetoesteróide, etiocolanolona, androsterona e
metabolitos androstanediol na mucosa GI.
T1/2: via Oral -> 30 min; Via IM ou transdérmica -> 10 a 100 min.
Derivados sintéticos de testosterona
Modificações estruturais -> Hormonas que evitassem o metabolismo de 1ª passagem.
Bloqueio do metabolismo do 17β-OH com substituintes em 17α -> Androgénios com
maior BD e duração de acção por via oral. Incluem a metiltestosterona e a fluoximesterona.
17α-Metiltestosterona
Presença de um alquilo: reduz o metabolismo oxidativo hepático → aumenta BD oral.
Após adminstração oral é bem absorvida no TGI. Mais activa por via sublingual.
Possui as actividades androgénicas e anabólicas da testosterona.
Fluoximesterona
Substituiu-se um 9-flúor num análogo da 17α-metiltestosterona (tem 20x a actividade
anabolizante e 10x a androgénica desta).
Menos de 5% é excretada inalterada. Provoca retenção de sódio e água→ edema
REA dos androgénios esteróides:
Requisito para a actividade androgénica: esqueleto esteróide.
Grupos funcionais de O em 3 e 17 -> Não necessários, o núcleo base 5α-androstano já
tem actividade androgénica.
Derivados de eticolano com H em 5β -> Proporciona uma junção Cis aos anéis A/B ->
Não activos anabólica ou androginicamente.
Expansão dos anéis formando homo-derivados por inserção de um metileno num dos
anéis do núcleo esteróide ou contracção por remoção do metileno -> Reduz ou destrói
as actividades.
Introdução de um 3-ceto ou 3α-OH: aumenta actividade androgénica.
Introdução de um 17-ceto elimina a actividade.
OH em 17α não contribui para as actividades.

PFFH – Química Farmacêutica – UP7
57
Nenhum substituinte se aproxima da efectividade de um 17β-OH
Ésteres de acção-longa de compostos com 17β-OH são hidrolisados in vivo ao álcool
livre, activo -> Oxigénio nesta posição é importante para a ligação ao receptor.
17α-alquilo -> Previne mudanças metabólicas nesta posição -> Activos oralmente
Aumento do tamanho da cadeia alquilo em 17α -> Diminui a actividade
Incorporação de outros substituintes, como:
17α-etinil -> Compostos com actividade progestacional útil
(ex: etisterona)
Introdução de um isoxazole à etisterona -> Danazol.
O Danazol é potente anti-gonadotrópico. Fraca actividade
androgénica e anabólica. Metabolizado pelo CYP3A4 a 2-
hidroximetiletisterona, inactiva.
Várias modificações na 17α-metiltestosterona -> Compostos anabólicos activos VO. 2
análogos hidroxilados: oximesterona e oximetolona -> 3x a actividade anabólica e
metade da androgénica da testosterona
Substituição halogénica: menor actividade excepto em 4 e 9 (ex: fluoromesterona)
Substituição do C em 2 por O -> Único esteróide heterocíclico com sucesso dentro
de vários azasteróides e oxasteróides. Alguns dos 2-oxasteróides são potentes
agentes anabólicos
C sp2 hibridizado no anel A -> Mais plano -> Aumenta actividade anabólica dando:
19-noresteróides:
Taxa favorável de actividade anabólica em relação à androgínica.
Substituições em 1, 2, 7, 17 e 18 -> Actividade favorável na clínica.
- Para os agentes anabólicos, conhecer as estruturas e as características estruturais mais
relevantes na actividade anabólica.
Não é possível dissociar os efeitos anabólicos
dos androgénicos -> Muitas acções dos esteróides
anabolizantes são semelhantes às dos androgénios.
A desidrogenação da 17α-metiltestosterona
com dióxido de selénio -> Análogo 1,4-dieno ->
metandrostenolona com maior acção anabólica e
menor androgénica.

PFFH – Química Farmacêutica – UP7
58
Oxandrolona: Análogo 2-oxasteróide da 17α-metiltestosterona, com uma lactona no anel A e
é susceptível a hidrólise in vivo. 3x mais actividade anabólica que 17α-metiltestosterona, leves
propriedades androgénicas.
Stanozolol: pirazole heterocíclico usado pelos efeitos anabólicos
Testolactona
18-oxasteróide, análogo da D-homo-oxoandrostandienediona com anel D que é uma
lactona de 6 membros
Alquilação nas posições 1, 2, 7 e 18 no androstano -> Aumenta actividade anabólica.
Acetato de metenolona -> Potente anabólico sem substituinte alquilo em 17α
Acetato de clortestosterona -> Agente anabólico halogenado
Androgénios sem metilo em 10 do núcleo esteróide -> Importante classe de anabólicos
-> 19-norandrogénios
Remoção do 19-CH3 -> Diminuição da actividade androgénica, retenção da anabólica
Estes esteróides podem ser sintetizados por redução de Birch do anel A aromático de
um estrogénio 3-metoxi a um 2,5(10)-estradieno
Clivagem do éter enol com HCl -> Derivado 19-nortestosterona -> Separação da
actividade anabólica da androgénica -> Noretandrolona (análogo 17α-etil) e
etilestrenol (mais potente).
Fenopropionato e decanoato de nandrolona
Ésteres da 19-nortestosterona, após lenta hidrólise in vivo liberta 19-nortestosterona
Decanoato de nandrolona -> Acção mais longa.
3.2.2 Androgénios Não-Esteróides - Conhecer as estruturas, características e REA dos
moduladores selectivos dos receptores de androgénios.
Modulador selectivo dos receptores de androgénios (SARM) -> Antagonista ou
agonista fraco na próstata e potente agonista no osso e músculo, disponível por VO com baixa
hepatotoxicidade. Farmacóforos dos SARM, 4 categorias:
1) N-arilpropionamidas;
2) Hidantoínas bicíclicas;
3) Quinolinas tricíclicas;
4) Análogos das tetrahidroquinolinas;
Estes androgénios não-esteróides não são
substratos da aromatase nem da 5α-redutase, mas têm

PFFH – Química Farmacêutica – UP7
59
afinidade como agonistas totais nos receptores de androgénios nos órgãos anabólicos
(músculo e osso); ou como parciais nos tecidos androgénicos (próstata e vesículas seminais).
REA das N-arilpropionamidas
Pesquisa focada em análogos de N-arilpropionamida, usando os elementos estruturais
chave da bicalutamida - antagonista androgénico
Uma série de análogos da bicalutamida, com grupos atractores de e- (ciano ou nitro
em 4 e trifluorometil em 3) no anel A e com flúor ou acetilamino em para no anel B
(R2), demonstraram afinidade de ligação aos receptores de androgénios in vitro
Os isómeros R, que com trifluorometil em vez de metilo em R2 têm maior afinidade e
actividade que os isómeros S.
o Os análogos sulfido (substituição do oxigénio do éter por um enxofre) têm maior
afinidade para os receptores, no entanto, a oxidação hepática da ligação de enxofre
leva a rápida inactivação in vivo → diminui a eficácia
Actividade agonista parcial: posição 3 do anel A está substituída por um trifluorometil.
Substituindo o anel A por um heterocíclico a actividade não é retida, provavelmente
devido ao impedimento estereoquímico na ligação ao receptor, no entanto, um anel B
heterocíclico retém a afinidade para o receptor
Porções electrotractoras pequenas em R2, como F, Cl, NO2 ou CN: Óptimos.
Pelo nitro aromático estar associado à toxicidade hepática, foi substituído por um
electroatractor não redutível (ex: CN) -> SARM mais potente e eficaz com FK favorável.
Diferenças mínimas na estrutura do ligando podem levar a agonistas ou antagonistas
Um agonista parcial ou total é influenciado pela conformação estereoisomérica, assim
como pelos efeitos estéricos e electrónicos dos substituintes.
3.3. Antagonistas de androgénios
3.3.1. Antagonistas dos receptores de androgénios - esteróides e não-esteróides
- Estruturas e principais características
Flutamida, Nilutamida, Bicalutamida -> Antiandrogénios
puros pois apenas se ligam aos receptores de androgénios.
São não-esteróides.
Têm vantagens sobre os antiandrogénios esteróides,
como o acetato de megesterol ou de ciproterona, em
termos de especificidade, selectividade e em FK.
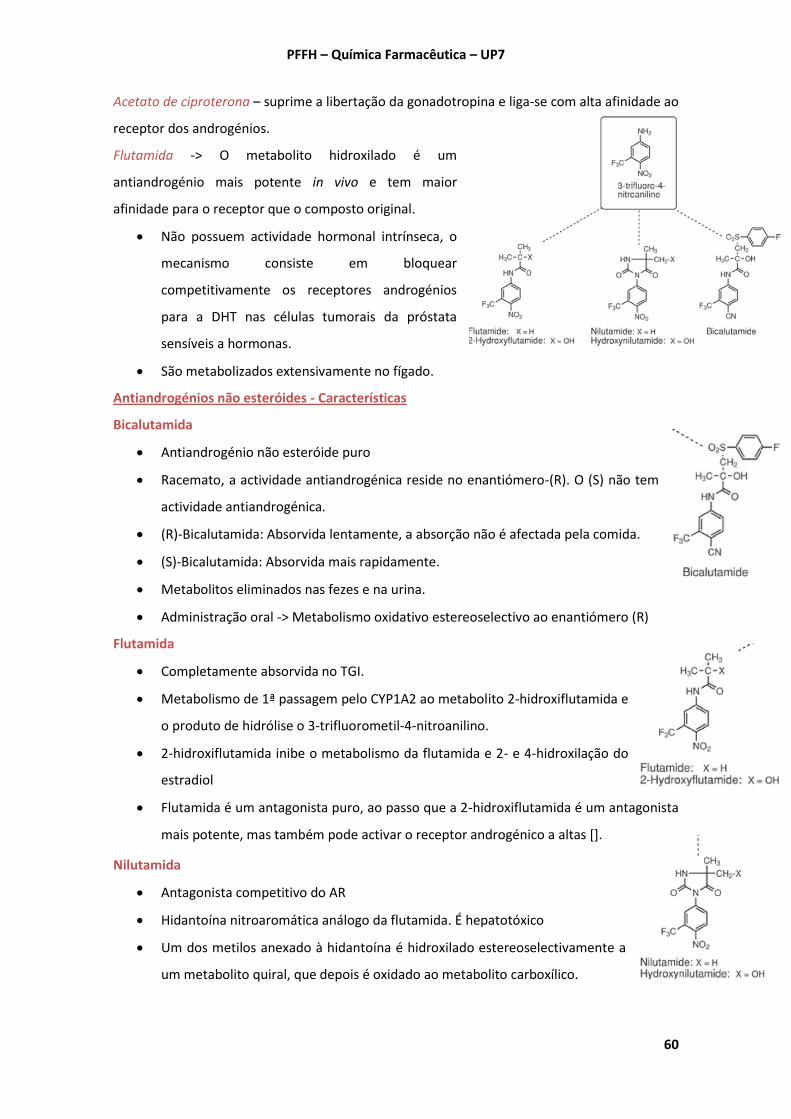
PFFH – Química Farmacêutica – UP7
60
Acetato de ciproterona – suprime a libertação da gonadotropina e liga-se com alta afinidade ao
receptor dos androgénios.
Flutamida -> O metabolito hidroxilado é um
antiandrogénio mais potente in vivo e tem maior
afinidade para o receptor que o composto original.
Não possuem actividade hormonal intrínseca, o
mecanismo consiste em bloquear
competitivamente os receptores androgénios
para a DHT nas células tumorais da próstata
sensíveis a hormonas.
São metabolizados extensivamente no fígado.
Antiandrogénios não esteróides - Características
Bicalutamida
Antiandrogénio não esteróide puro
Racemato, a actividade antiandrogénica reside no enantiómero-(R). O (S) não tem
actividade antiandrogénica.
(R)-Bicalutamida: Absorvida lentamente, a absorção não é afectada pela comida.
(S)-Bicalutamida: Absorvida mais rapidamente.
Metabolitos eliminados nas fezes e na urina.
Administração oral -> Metabolismo oxidativo estereoselectivo ao enantiómero (R)
Flutamida
Completamente absorvida no TGI.
Metabolismo de 1ª passagem pelo CYP1A2 ao metabolito 2-hidroxiflutamida e
o produto de hidrólise o 3-trifluorometil-4-nitroanilino.
2-hidroxiflutamida inibe o metabolismo da flutamida e 2- e 4-hidroxilação do
estradiol
Flutamida é um antagonista puro, ao passo que a 2-hidroxiflutamida é um antagonista
mais potente, mas também pode activar o receptor androgénico a altas [].
Nilutamida
Antagonista competitivo do AR
Hidantoína nitroaromática análogo da flutamida. É hepatotóxico
Um dos metilos anexado à hidantoína é hidroxilado estereoselectivamente a
um metabolito quiral, que depois é oxidado ao metabolito carboxílico.

PFFH – Química Farmacêutica – UP7
61
In vitro, o nitro é reduzido a amina e hidroxilaminas. A redução é catalisada pela NO
sintase por via da formação de um ou ambos radicais livres de aniões nitro ou a
redução a derivados hidroxilamina podem explicar alguns efeitos tóxicos do fármaco.
NO sintase está envolvida na formação de derivados reactivos de NO e espécies de
oxigénio na interacção com alguns compostos xenobióticos.
3.3.2. Inibidores da biossíntese de androgénios
Existe relação entre a exposição à testosterona e DHT e o risco de desenvolver cancro
da próstata -> Prevenção com inibidores específicos de enzimas chave associadas à
biossíntese de androgénios -> 2 enzimas fundamentais são consideradas para inibição:
5α-redutase -> inibe a formação de DHT.
17α-hidroxilase/17,20-liase -> inibe a biossíntese de testosterona.
- Conhecer as estruturas e características dos inibidores da 5α-redutase e saber explicar o
seu mecanismo de actuação.
Mecanismo de acção
Inibidores da biossíntese de DHT -> Menor [DHT], bloqueando a acção androgénica.
Enzima crítica para a inibição: 5α-redutase (converte a testosterona em DHT).
1º agente que demonstra esta inibição foi um análogo da progestina, medrogesterona.
2 derivados “azasteroid-17-amide” da medrogesterona ->
Inibidores irreversíveis:
Finasteride: inibidor selectivo
Dutasterida: Inibidor não selectivo
A inibição do tipo 2 da 5α-redutase suprime o
metabolismo da testosterona a DHT, resultando numa
diminuição de DHT no plasma e na próstata.
Finasterida é 30x mais selectiva para o tipo 2 da 5α-redutase e a Dutasterida é 10x
mais potente como inibidor para o tipo 2 da 5α- redutase do que para o tipo 1.
Interacções farmacológicas
Finasterida e a Dutasterida: metabolizadas pelo CYP3A4 -> Inibidores do CYP3A4
podem aumentar os níveis do fármaco e causar interacções.
Finasteride
Inibidor selectivo do tipo 2 da 5α-redutase
Suprime cerca de 70% dos níveis de DHT no plasma e de 85-90% na próstata. A
restante na próstata resulta provavelmente do tipo 1 da 5α-redutase

PFFH – Química Farmacêutica – UP7
62
Não tem afinidade para os AR nem efeitos androgénicos, antiandrogénicos,
estrogénicos, antiestrogénicos ou progestacionais.
Acção inibitória da finasterida: similaridade na estrutura com a testosterona -> Liga à
enzima e é reduzido a
dihidrofinasterida no lugar da
testosterona. Capacidade de
agir por um mecanismo,
durante o qual “prende” o
NADPH, formando um aducto
covalente NADP-
dihidrofinasterida. Este
liberta muito lentamente
dihidrofinasterida com meia-
vida de um mês.
BDoral ~65%. Absorção não afectada pela comida
90% da Finasterida em circulação encontra-
se ligada a proteínas plasmáticas
Consegue atravessar a BHE.
Extensamente metabolizada no fígado, pelo
CYP3A4 originando 2 metabolitos: monohidroxilfinasterida posteriormente
metabolizada através de um aldeído intermediário originando o 2º metabolito, ácido
monocarboxílico.
Metabolitos apresentam 20% da inibição da Finasterida para a 5α- redutase
40% Excretado na urina e cerca de 57% nas fezes
Dutasterida
Inibidor competitivo do tipo 1 e 2 da 5α-redutase ->
melhor e mais consistente redução do DHT no plasma
Extensivamente metabolizado pelo CYP3A4 em:
4’-hidroxidutasterida
1,2- dihidrodutasterida (menos potente)
6’-hidroxidutasteride (comparado com o fármaco
original, inibidor do tipo 1 e 2 da 5α-redutase)
- Conhecer as estruturas e principais características dos inibidores da 17α-hidroxilase e
17,20-liase

PFFH – Química Farmacêutica – UP7
63
A 17α-hidroxilase/17,20-liase converte a pregnenolona a DHT e depois a testosterona.
Como a testosterona tem actividade androgénica, a inibição da sua biossíntese é útil
em doenças dependentes de androgénios (ex: cancro da prostata).
Inibidores desta podem prevenir a biossíntese dos androgénios.
O agente antifúgico Ketoconazole é um inibidor da 17α-hidroxilase
O imidazole não esteróide (Liarozole), encontra-se em
desenvolvimento como potencial inibidor selectivo da 17α-
hidroxilase/17,20-liase.
Abiraterona e Sa40 são análogos heterocíclicos do MDL 27,302 e
potentes inibidores irreversíveis da 17α-hidroxilase. Necessárias duplas
em 16 e 17 para se formarem ligações irreversíveis entre os esteróides
17-piridil e 17-pirimidil para o 17αhidroxilase.
Oxidação a epóxidos não se encontra -> epóxido é inibidor fraco.
- Conhecer outros inibidores da biossíntese de androgénios
Leuprolide, goserelin e buserelina suprimem secreção de
LH e síntese da testosterona
Agonistas LHRH suprimem a síntese de DHT intraprostática
e de testosterona -> Efeito não imediato, no entanto,
ocorre 2-4 semanas após o início da terapia.
F. Homeostase do Cálcio
1. Introdução
- Conhecer a estrutura geral da calcitonina e da hormona paratiróide
Calcitonina (CT)
Péptido de 32 AA sintetizado nas células parafoliculares C da
tiróide; Igual em 7 dos 9 primeiros AA, na glicina 28 e
prolilamida no C-terminal em diferentes espécies.
Amida da prolina C-terminal (Pro-NH2) e ponte dissulfito entre
Cys em 1 e 7-> Importantes.
CT dos peixes é 20-25x mais potente que a dos mamíferos pela maior afinidade para
receptores nos ossos e rins.
Toda a sequência de AA é requerida para a actividade biológica;
Pró-calcitonina: Precursor. Facilita transporte para dentro da célula e secreção CT;
Os resíduos 10-27 podem ser alterados, para alterar a potência e t1/2 do análogo resultante.
Hormona Paratiróide - PTH

PFFH – Química Farmacêutica – UP7
64
Cadeia única com 34 a.a., com 2 precursores durante a biossíntese:
pré-pró-PTH (115AA) – RER da paratiróide, clivada a:
pró-PTH (84AA) no espaço cisternal do RE.
A hormona activa é finalmente produzida (34AA) no complexo de Golgi e armazenada em
grânulos secretores da paratiróide;
- Estrutura das diferentes formas da vitamina D, biossíntese e activação,
requisitos importantes para a actividade e alterações para a aumentar.
A Vit. D (calciferol) pode encontrar-se em óleos de peixes e leite.
Vitamina D
Deriva do colesterol. Sintetizada da pro-hormona Colecalfiferol (D3), produto da radiação
solar UV do 7-desidrocolesterol na pele.
A Vit. D1 obtém-se do Ergosterol (pré-vitamina D2)-> Rearranjo foto-oxidativo ->
Taquisterol + Complexo de Ergocalciferol (D2 antirraquitico) e Luminesterol (inactivo).
Biossíntese da vitamina D2:
1. Ruptura do anel B do ergosterol por radiação UV.
2. A ruptura da ligação σ entre os C(9) e C(10) é acompanhada pela
formação de uma nova dupla entre os C(6) e C(7) em cis.
3. Reordenação de sistema de 3 duplas conjugadas por transposição
sigmatrópica [1,7].
4. Isomerização a s-trans (ligação conjugada de carácter duplo C6-
C7-> Apesar de simples, devido à conjugação, tem carácter parcial
de dupla, e por isso restrição rotacional)
• O 7-desidrocolesterol, do metabolismo do colesterol no humano,
transforma-se por radiação, em colecalciferol.
Vit. D3: Sofre activação no fígado ou rim por 2 oxidações a metabolitos mais polares:
1. a 25-hidroxicolecalciferol-> Microssomas de hepatócitos pela Vit.
D25-hidroxilase. Esta não é regulado pelas [Ca] plasmáticas.
2. Activada em resposta a hipocalcémia e secreção de PTH;
Mitocôndrias renais; Vit. D 1α-
hidroxilase. O produto – 1,25-
dihidroxicolecalciferol (calcitriol) - é a
forma activa da Vit. D. A biossíntese de
vitamina D é regulada pelas [Ca, Fosfato,
PTH e Vit. D Activa] plasmáticas.

PFFH – Química Farmacêutica – UP7
65
• A acção de uma hidroxilase renal estimulada pela PTH introduz
um OH em 24->Inactivação.
• As reacções fotoquímicas abrem o anel B nos sistemas 5,7-didesidrogenados (ex: 7-
desidrocolesterol e ergosterol) são exemplos de reacções electrocíclicas.
• Estas com as reacções de cicloadição e as transposições sigmatrópicas são reacções
pericíclicas, nas quais as ligações se rompem e formam em simultâneo.
• A formação da D2 e D3 requer transposição sigmatrópica [1,7] do composto A, no qual um H
do CH3 em C(19) passa para C(9) do esteróide, originando o composto B.
• A geometria s-cis da ligação C(6)-C(7) no trieno B corresponde à configuração cis no trieno
A, formado na reacção de electrociclação.
A terminologia s-cis indica uma configuração sinperiplanar em torno da ligação dupla.
A mudança para configuração antiperiplanar (s-trans) conduz, por fim, às vitaminas D.
Ambas as formas referidas da vitamina D têm acção similar no
organismo humano.
A 1,25-dihidroxivitamina é transportada para o osso, intestino e
outros, podendo ser considerada uma hormona.
Este composto actua nos receptores nucleares controlando níveis de Ca e fosfato,
promovendo a mineralização do osso. Hipoparatiroidismo e osteoporose pós-menopausa.
OH-1 e 25: Críticos para a interacção. Eliminação: Reduz afinidade 1000x.
3 OH do calcitriol desempenham papel
importante.
Núcleo das células-> Receptores
específicos para 1,25-
dihidroxicolecalciferol *1α,25(OH)2D3].
A interacção estimula a produção de
proteínas, como a osteocalcina.
Modificações na cadeia em C-17
produzem compostos com maior
actividade que o natural. Esta é muito
flexível-> OH em C-25 pode situar-se
numa região do espaço muito ampla.
O epímero em C-20 (2) é muito mais
potente que a vitamina natural.

PFFH – Química Farmacêutica – UP7
66
A afinidade pelo receptor aumenta na ordem 6<3<4<5, o que corresponde a uma
localização do hidroxilo C-25 de 1 em uma das 4 regiões referidas, diferentes das do
epímero 2.
Resumo, as modificações que aumentam a potência são:
Epimerização em C-20;
Substituição do metileno em C-22 por oxigénio (7);
Introdução de ligação dupla ou tripla na cadeia lateral (8) ou na posição C-16 (9);
Eliminação do metilo em C-18 (11);
Alargamento da cadeia (12);
Metilação dos grupos metilo terminais C-26 e C-27 (14);
Metilação em C-20 (17).
Regulação do metabolismo global do cálcio:
O metabolismo global do cálcio é regulado pela PTH e pela CT.
A PTH, bem como o estado hipocalcémico, activam a enzima
1α-hidroxilase renal, que leva à formação da vitamina D3 activa [1,25-(OH)2D3].
[Ca, Calcitriol e PTH] elevadas-> Activada uma 24-hidroxilase renal, que inactiva a Vit. D3. Os
derivados 1,24,25-triol e 24,25-diol também suprimem a secreção de PTH.
2. Terapias farmacológicas da Osteoporose
2.1- Análogos de estrogénios e moduladores selectivos dos receptores de estrogénios
- Estruturas e as semelhanças/diferenças entre elas, requisitos estruturais para a actividade.
REA
Únicos anti-estrogénios puros: Estrogénios 7α substituídos.
Na classe dos agentes triariletileno, o anel A é fundamental para ocorrer a ligação ao
ER pois mimetiza o 3-fenólico essencial dos estrogénios.
Orientação dos 3 aril num arranjo em hélice: Importante para ligação ao receptor.
Raloxifeno
O raloxifene hidrocloreto, derivado benzotiofeno. Análogo semi-
rígico do tamozifeno
Ambos têm actividade agonista em certos tecidos (ex: ossos-
osteoblastos e osteoclastos- cardiovascular) e actividade antagonista em outros (ex: útero)
Farmacocinética
Rapidamente absorvido VO. Baixa BD por metabolismo de fase II.
Metabolitos excretados pela bílis com reciclagem enterohepática.

PFFH – Química Farmacêutica – UP7
67
Ocorre no intestino e consiste em glucoronidação catalisada
pela uridina difosfato glucoronosiltransferase (UGT)
Refluxo do glucoronido pela gp-P (responsável por baixa BD)
2.2- Bifosfonatos
- Estrutura-base e mecanismo de actuação.
Mimetizam o pirofosfato. O O no P-O-P é substituído por um C-> Base não-hidrolisável.
Mecanismo de acção: ligam-se selectivamente à porção
hidroxiapatite do osso, diminuindo o nº de locais ao longo da
superfície óssea em que a reabsorção óssea mediada por
osteoclastos pode ocorrer-> Osteoblastos formam novo osso bem
mineralizado com competição inferior dos osteoclastos.
- Diferentes bifosfonatos, classificação química, REA e FK; principais características.
Ácido alendrónico e ácido ibandrónico (+importantes)
REA
C central do fosfonato germinal-> Susbtituído com
grande variedade de grupos para dar origem a uma
família de compostos com propriedades FQ e
actividades diferentes.
Um OH (R1) maximiza a afinidade do fármaco pela
hidroxiapatite assim como as propriedades anti-reabsorção do mesmo.
O R2 varia e tem importância na potência desta classe de compostos.
Os bifosfonatos amino em R2 são mais potentes do que o etidronato e clodronato.
A cadeia linear de 4C’s em R2 do alendronato é mais potente que o de 3C’s do pamidronato
ou o de 6C’s do neridronato.
Alquilação da amina-> Aumento da potência, demonstrado com aminas ramificadas em R2
ou naqueles contendo anéis em R2.
Os de 3ª-geração contêm cadeia lateral heterocíclica básica em R2 ligada ao C central por
uma variedade de ligações (potência: NH > CH2 > S > O).
R2 interage num “sítio activo” e participa numa interacção molecular específica.
Bifosfonato e o OH em R1 devem ser considerados essenciais.
Farmacocinética
Absorção (1-5%) e penetração celular bastante fraca devido à natureza polar.
50% da dose absorvida é “capturada” pelo osso em 4-6 horas. Restante: Excretada nos rins.
Fármaco captado pelo osso concentra-se em locais a sofrer remodelação activa.

PFFH – Química Farmacêutica – UP7
68
Pequeno T1/2 em circulação.
Apenas se libertam do osso quando este é reabsorvido-> T1/2 tecidual de 1-10 anos. Apenas
se mantêm activos quando expostos em superfícies de reabsorção do osso.
Agentes Específicos
ALENDRONATO DE SÓDIO:
2ªgeração, 1º aprovado pela FDA para a osteoporose. 1000x mais potente que etidronato.
Efeitos 2ºs: Esofagite química por pouca ingestão de água do doente se deitar após a toma.
Para aumentar a absorção, suplementos de Ca ou antiácidos contendo Al ou Mg-> Tomados
IBANDRONATO DE SÓDIO:
BDoral é baixa (0,6 %). Afectada pela comida, e suplementos de Ca e Vit. D ou antiácidos.
Pela maior quantidade de cálcio na água mineral, os doentes não a devem consumir.
Fármacos que inibam a secreção gástrica aumentam a absorção do ácido ibandrónico.
Não metabolizado. Quando não ligado ao osso: Eeliminado via renal.
2.3 Calcitonina
– Origem, semelhanças/diferenças e vantagens /desvantagens da calcitonina de salmão.
A CT isolada de peixes é mais potente que a dos mamíferos
pela afinidade superior para receptores ósseos e renais. A
CT de salmão difere da humana em 16AA e está disponível
na forma de spray nasal e injectável.
2.4 Outros agentes relevantes
SAIS DE CÁLCIO INORGÂNICOS
Ingestão de sais de Ca2+ nas fases de crescimento (1200-1500 mg/dia), aumenta
a densidade mineral óssea e reduz o risco de desenvolvimento de osteoporose.
G. Disfunção eréctil
1. Inibidores da fosfodiesterase V
- Estruturas dos nucleótidos lineares e cíclicos e compreender as interconversões
enzimáticas no organismo e as enzimas envolvidas.
Os nucleótidos cíclicos adenosina 3’,5’-monofosfatase
cíclica (cAMP) e a guanosina 3’,5’-monofosfatase cíclica
(cGMP)-> Papel na mediação de sinais celulares em
resposta a hormonas, NT e citocinas.
Estes iniciam-se por ligação de uma hormona ao receptor
da proteína G, com activação das adenilil e guanilil ciclases
que sintetizam cAMP e cGMP, respectivamente.

PFFH – Química Farmacêutica – UP7
69
Por sua vez, estes novos nucleótidos sintetizados activam efectores, cinases A e G que
fosforilam substratos como canais iónicos e factores de transcrição. Os sinais podem levar a
alterações na expressão do gene, metabolismo e na regulação de uma variedade de
funções celulares (ex. resposta imune).
As fosfodiesterases (PDEs) hidrolisam nucleótidos cíclicos às formas lineares inactivas (5’-
AMP e 5’-GMP) sendo reguladores negativos da sinalização de nucleótidos cíclicos.
- Estrutura das fosfodiesterases de nucleótidos cíclicos, especificidade, incidindo na PDE5.
PDEs: Hidrólise do diéster 3’-fosfato de nucleótidos cíclicos dando monoéster 5’-fosfato
acíclico. Distinguidas pela estrutura 1ª, distribuição nos tecidos, localização intracelular,
regulação e especificidade:
Famílias 4, 7 e 8 hidrolisam cAMP;
Famílias 5, 6 e 9 hidrolisam cGMP;
Famílias 1, 2, 10 e 11 hidrolisam
ambos os nucleótidos cíclicos.
PDEs humanas: Domínio catalítico C-terminal e vários domínios
de transdução de sinal que devem ser sensores de sinais
intracelulares que levam a alterações na conformação da PDE e à sua actividade.
O domínio catalítico contém um núcleo de 16 α-hélices num arranjo compacto de 3
domínios compostos por aH1-7, aH8-11, e aH12-16, respectivamente.
O sítio activo na intersecção destes três domínios forma uma bolsa hidrofóbica profunda
com uma abertura estreita e um amplo espaço interno.
11 dos 16AA estão presentes no sítio activo-> Reconhecimento e catálise por 4 interacções:
1. Ligação a dois iões metálicos divalentes;
2. Coordenação da mistura diéster fosfato;
3. “Aperto” hidrofóbico do anel de
nucleótidos altamente conservados por AA
hidrofóbicos que compactam o substrato
no local activo;
4. Rede de pontes de H que ajudam a determinar a especificidade de nucleótidos.
Tratamento da disfunção eréctil-> Inibidores competitiva da PDE5 (ex: sildenafil, vardenafil
e tadalafil) actua por reforço e prolongamento do relaxamento do músculo liso e
vasodilatação como resultado da estimulação sexual.
Estimulação sexual: Libertação de NO pelas células endoteliais bem como dos nervos
cavernosos, activando a guanilato ciclase-> Estimula produção de cGMP-> Diminuição dos
níveis de Ca celulares, relaxamento das células do ML dos corpos cavernosos e à erecção.

PFFH – Química Farmacêutica – UP7
70
Pela degradação do cGMP ser catalizada pela PDE5, a inibição desta leva a níveis elevados
de cGMP nos corpos cavernosos-> Melhora capacidade para manter uma erecção.
- Estruturas e características dos inibidores da PDE5, especificidade de inibição e REA
Primeiro: Teofilina. Esta e outras xantinas (Ex. 3-isobutil-1-
metilxantina – IBMX) têm estrutura planar que mimetiza a planaridade das
bases de cAMP e cGMP. São não selectivas, contudo, adoptam modelos de
ligação múltiplos no sítio activo das PDEs.
O sildenafil e vardenafil são análogos de azaguanina substituídos com
pirazolopirimidona que mimetiza o modo de ligação da guanina no cGMP,
enquanto que o 3’-n-propil liga-se numa região similar à ribose.
A selectividade PDE5\PDE do sildenafil e vardenafil parece girar em torno da
glutamina (Gln-87), que interactua com a pirimidina do cGMP na conformação
de guanina selectiva.
Maior especificidade resulta da alquilpiperazina, que não é sobreponível com
o nucleótido e é exposto à superfície da proteína por abertura do sítio activo. As
interacções com AA hidrofóbicos circundantes não são encontradas em regiões
equivalentes da PDE4-> Pode contribuir para a especificidade.
Sildenafil e o vardenafil têm boa selectividade PDE5\PDE contra PDEs
selectivas de cAMP (PDE 4, 7 e 8) mas vão inibir outras PDEs selectivas de cGMP (1,
6, 9, 10 e 11). A sua baixa selectividade PDE5\PDE6 é um caso particular que se
pensa responsável por certos efeitos adversos na visão.
A falta de selectividade pode ser explicada pela homologia entre sítios activos
da PDE5 e PDE6.
A elevada selectividade PDE5 e PDE5\PDE3 do sildenafil e vardenafil é desconhecida.
SILDENAFIL
É rapidamente absorvido. Tem 96% de ligação às proteínas plasmáticas.
É metabolizado pela CYP3A4
O metabolito N-desmetilsildenafil possui 50% da actividade da molécula parental
REA
• Diferenças entre sildenafil, vardenafil e tadalafil: Heterocíclico usado para mimetizar a
purina do cGMP
• Embora o heterocíclico e o N-substituinte (etil versus metil) ligados à cadeia lateral
piperazina sejam a diferença entre o sildenafil e o vardenafil, esta não explica o facto do
vardenafil ter potência 20x maior para a inibição da PDE5 que o sildenafil.

PFFH – Química Farmacêutica – UP7
71
• O metil/etil na piperazina têm papel pequeno na diferença de potência,
e as diferenças no heterocíclico têm um papel crítico para a alta
potência do vardenafil.
2. Outros – Estruturas e características
2.1. Alprostadilo - Prostaglandina E1
Combinação de PGE1, papaverina e fentolamina é usada como injecção
intracavernosal na DE.
A PGE1 produz endogenamente relaxamento vascular do ML e causa
vasodilatação por activação da via adenilato ciclase/cAMP.
Por injecção intracavernosal ou supositório intrauretral actua para
relaxar o ML do corpo cavernoso e as artérias cavernosas.
O inchaço, alongamento e rigidez do pénis resulta quando o fluxo arterial atinge
rapidamente o corpo cavernoso para expandir os espaços lacunares
Metabolizado na uretra, próstata e corpo cavernoso a ácido 7-hidroxi-
5,11-dicetotetranorprosta-1,16-dióico e ácido 5,7-dihidro-11-
cetotetranorprostano-1,16-dióico
A maior rota de excreção dos seus metabolitos é renal.
Se absorvido sistemicamente é metabolizado num único passo nos pulmões
2.2. Papaverina e fentolamina
PAPAVERINA (Injecção Intracavernosa)
Em combinação com fentolamina para facilitar erecções na DE. Útil em doentes com DE
orgânica; Menos útil na DE por problemas endócrinos ou medicamentos.
Fraco e não específico inibidor da PDE-> relaxamento do ML cavernoso e vasodilatação das
artérias penianas-> Aumento do fluxo sanguíneo arterial-> Dilatação e alongamento pénis.
Descarga venosa reduzida por aumento da resistência venosa.
Libertada lentamente na circulação venosa com mínimos efeitos sistémicos.
Pico de acção em 10min e duração de 1-6h com administração com fentolamina.
FENTOLAMINA (intracavernoso)
Antagonismo α1e 2-> Vasodilatação e redução das resistências periféricas
Causa relaxamento dos ML cavernosos e vasodilatação das artérias do pénis. Descarga
venosa reduzida por aumento da resistência venosa
É lentamente libertada na circulação venosa, com mínimos, ou nenhuns efeitos sistémicos.


















