
Classificação das dispersões coloidais Dupla camada...
30
Classificação das dispersões coloidais Dupla camada elétrica: Equação de Lippman e modelos Estabilidade e coagulação de dispersões coloidais Ahmad Hassan Mourad - 9877784 Igor Rodrigues Arangues - 9774960 Larissa de Camargo Sousa - 9862210 Luiz Galvão de Almeida Passos - 9359789 Murillo Giraldi Magioli Cadan - 9002572 Vinicius Hideki Uehara - 8914417 Lorena - SP 2018
Transcript of Classificação das dispersões coloidais Dupla camada...

Classificação das dispersões coloidais
Dupla camada elétrica: Equação de Lippman e modelos
Estabilidade e coagulação de dispersões coloidais
Ahmad Hassan Mourad - 9877784
Igor Rodrigues Arangues - 9774960
Larissa de Camargo Sousa - 9862210
Luiz Galvão de Almeida Passos - 9359789
Murillo Giraldi Magioli Cadan - 9002572
Vinicius Hideki Uehara - 8914417
Lorena - SP
2018

SUMÁRIO 1. INTRODUÇÃO ..............................................................................................3 2. DISPERSÕES ...............................................................................................3
2.1. COLOIDES OU DISPERSÕES COLOIDAIS .....................................3
2.2. PROPRIEDADES DOS COLOIDES ..................................................4
2.3. INTERAÇÃO ENTRE PARTÍCULAS COLOIDAIS ............................6
3. SISTEMAS COLOIDAIS ...............................................................................8
3.1. GEL ....................................................................................................8
3.2. SOL ....................................................................................................9
3.3. ESPUMA ..........................................................................................10
3.4. EMULSÃO .......................................................................................11
4. DUPLA CAMADA ELÉTRICA ....................................................................12
5. ELETROCAPILARIDADE E EQUAÇÃO DE LIPPMAN .............................14
6. MODELOS DE DUPLA CAMADA ..............................................................16
6.1. MODELO DE HELMHOLTZ (1853) .................................................16
6.2. MODELO DE GOUY-CHAPMAN (1910 – 1913) .............................18
6.3. MODELO DE STERN (1924) ...........................................................19
6.4. MODELO DE GRAHAME (1947) .....................................................20
7. ESTABILIDADE E COAGULAÇÃO DE DISPERSÕES COLOIDAIS ........21
7.1. ESTABILIDADE ...............................................................................21
7.1.1. MECANISMO ..............................................................................22
7.2. COAGULAÇÃO ................................................................................24
7.2.1. MECANISMOS ............................................................................26
7.2.1.1. COMPREENSÃO DA DUPLA CAMADA ELÉTRICA .....26
7.2.1.2. ADSORÇÃO E NEUTRALIZAÇÃO .................................27
7.2.1.3. VARREDURA ...................................................................27
7.2.1.4. ADSORÇÃO E FORMAÇÃO DE PONTES .....................28
8. REFERÊNCIAS ...........................................................................................29

1. INTRODUÇÃO
A química dos coloides está bastante relacionada com o dia-a-dia do
cidadão e os sistemas coloidais tanto são encontrados na natureza, nos reinos
mineral, vegetal e animal, como podem ser sintetizados para o bem-estar do
homem na forma de bens de consumo e para processos industriais que
propiciam melhores condições de vida. O estudo dos coloides também pode
ajudar a evitar a formação desses sistemas na natureza, quando poluem o ar
(fumaça), a água (esgoto doméstico e industrial) e os solos (resíduos sólidos).
Sendo assim, as respostas para muitas dessas questões ambientais são
encontradas através da química dos coloides.
2. DISPERSÕES
2.1. COLOIDES OU DISPERSÕES COLOIDAIS
Coloides são misturas heterogêneas de pelo menos duas fases
diferentes, com a matéria de uma das fases na forma finamente dividida
(sólido, líquido ou gás), denominada fase dispersa, misturada com a fase
contínua (sólido, líquido ou gás), denominada meio de dispersão. A ciência dos
coloides está relacionada com o estudo dos sistemas nos quais pelo menos um
dos componentes da mistura apresenta uma dimensão no intervalo de 1 a 1000
nanômetros (1 nm = 10-9 m).
Soluções de macromoléculas são misturas homogêneas e também são
consideradas coloides porque a dimensão das macromoléculas está no
intervalo de tamanho coloidal e, como tal, apresentam as propriedades
características dos coloides.
Os sistemas coloidais vêm sendo utilizados pelas civilizações desde os
primórdios da humanidade; os povos utilizaram géis de produtos naturais como
alimento, dispersões de argilas para fabricação de utensílios de cerâmica e
dispersões coloidais de pigmentos para decorar as paredes das cavernas com
motivos de animais e de caça.
Graham, em 1861, introduziu os termos: coloide e diálise em um estudo
sobre a difusão da matéria nos estados gasoso e líquido. Diálise é o processo
de separação através do qual, moléculas menores atravessam uma membrana

semipermeável enquanto as moléculas maiores ou partículas coloidais são
retidas pela mesma membrana e o termo coloide, do grego, significa cola e na
época referiu-se às soluções de goma arábica, que é substância sem estrutura
definida e de natureza viscosa, hoje conhecida como macromolécula. A goma
arábica (coloide) difundia mais lentamente que soluções de sais (cristaloide).
Sistemas coloidais estão presentes no cotidiano desde as primeiras
horas do dia, na higiene pessoal, como sabonete, xampu, pasta de dente e
espuma ou creme de barbear, e no café da manhã, leite, café, manteiga,
cremes vegetais e geleias de frutas. No caminho para o trabalho podemos
enfrentar neblina, poluição do ar. No almoço, temperos, cremes e maionese
para saladas. No entardecer, ao saborear cerveja, refrigerante ou sorvete
estamos ingerindo coloides. Os coloides ainda estão presentes em diversos
processos de produção de bens de consumo, incluindo o da água potável, os
processos de separação nas indústrias, de biotecnologia e de meio ambiente.
2.2. PROPRIEDADES DOS COLOIDES
Os princípios relacionados com os diferentes sistemas coloidais da
Tabela 1 baseiam-se em propriedades comuns a todos os coloides: tamanho e
elevada relação área/volume de partículas (Shaw, 1975). As partículas
dispersas podem ter tamanhos diferentes e por isso o sistema coloidal é
denominado polidisperso. Na prática, a maioria dos coloides obtidos pelo
homem é polidispersa. Os sistemas com partículas de um mesmo tamanho são
monodispersos. As macromoléculas de proteínas sintetizadas biologicamente
têm o mesmo tamanho e massa molecular, por isso dão origem a coloides
monodispersos. Diversos pesquisadores obtiveram coloides monodispersos de
polímeros sintéticos, de metais, de óxidos metálicos e de cloreto de prata.
Coloide Fase dispersa Fase de dispersão Exemplo
Aerossol líquido Líquido Gás Neblina, desodorante
Aerossol sólido Sólido Gás Fumaça, poeira
Espuma Gás Líquido Espuma de sabão
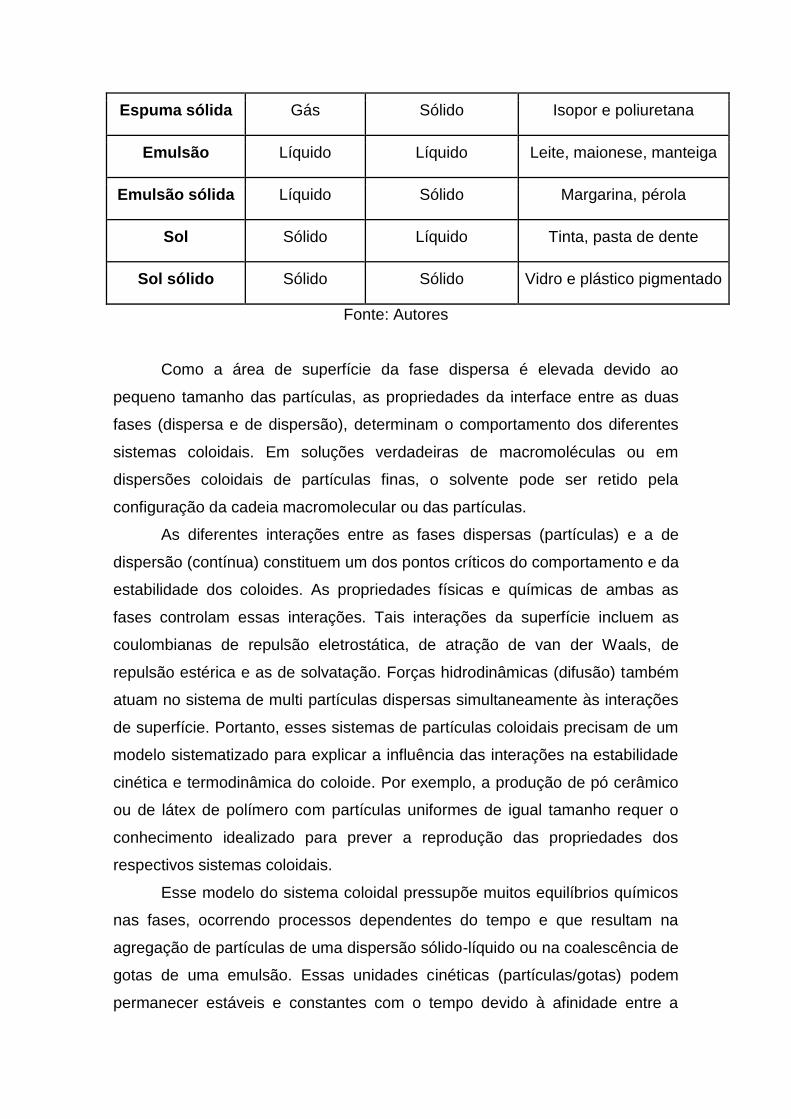
Espuma sólida Gás Sólido Isopor e poliuretana
Emulsão Líquido Líquido Leite, maionese, manteiga
Emulsão sólida Líquido Sólido Margarina, pérola
Sol Sólido Líquido Tinta, pasta de dente
Sol sólido Sólido Sólido Vidro e plástico pigmentado
Fonte: Autores
Como a área de superfície da fase dispersa é elevada devido ao
pequeno tamanho das partículas, as propriedades da interface entre as duas
fases (dispersa e de dispersão), determinam o comportamento dos diferentes
sistemas coloidais. Em soluções verdadeiras de macromoléculas ou em
dispersões coloidais de partículas finas, o solvente pode ser retido pela
configuração da cadeia macromolecular ou das partículas.
As diferentes interações entre as fases dispersas (partículas) e a de
dispersão (contínua) constituem um dos pontos críticos do comportamento e da
estabilidade dos coloides. As propriedades físicas e químicas de ambas as
fases controlam essas interações. Tais interações da superfície incluem as
coulombianas de repulsão eletrostática, de atração de van der Waals, de
repulsão estérica e as de solvatação. Forças hidrodinâmicas (difusão) também
atuam no sistema de multi partículas dispersas simultaneamente às interações
de superfície. Portanto, esses sistemas de partículas coloidais precisam de um
modelo sistematizado para explicar a influência das interações na estabilidade
cinética e termodinâmica do coloide. Por exemplo, a produção de pó cerâmico
ou de látex de polímero com partículas uniformes de igual tamanho requer o
conhecimento idealizado para prever a reprodução das propriedades dos
respectivos sistemas coloidais.
Esse modelo do sistema coloidal pressupõe muitos equilíbrios químicos
nas fases, ocorrendo processos dependentes do tempo e que resultam na
agregação de partículas de uma dispersão sólido-líquido ou na coalescência de
gotas de uma emulsão. Essas unidades cinéticas (partículas/gotas) podem
permanecer estáveis e constantes com o tempo devido à afinidade entre a

superfície da partícula e o solvente. Na ciência dos coloides, o sistema é
classicamente denominado coloide liofílico (do grego lyein = solvente + philein
= gostar de). No entanto, se as unidades cinéticas não permanecerem estáveis
com o tempo devido às interações entre elas e vierem a agregar-se, formarão
unidades maiores que se sedimentam sob a ação do campo gravitacional,
separando assim a fase dispersa da contínua. Esse sistema é também
classicamente conhecido como coloide liofóbico (repulsão ao solvente - phobos
= repelir).
Assim, existem coloides estáveis por muitos anos, enquanto em outros
as fases constituintes separam-se em pouco tempo. Por exemplo, os sóis de
ouro obtidos por Michael Faraday em 1864 permanecem estáveis até hoje e
estão expostos na Royal Society of Chemistry em Londres. Já a poeira
levantada pelo trânsito assenta rapidamente. As dispersões coloidais aquosas
são também sensíveis à presença de eletrólitos e de polieletrólitos (polímeros
carregados de alta massa molecular). As partículas coloidais podem agregar se
irreversivelmente na presença de eletrólitos e resultar em agregados grandes e
compactos (coágulos) por um processo denominado coagulação, enquanto na
presença de polieletrólitos pode haver a formação de agregados menos densos
(floculo), os quais podem ser facilmente rompidos e dispersos por agitação
vigorosa (cisalhamento). A coagulação do leite, por exemplo, resulta da adição
de vinagre (eletrólito) e a eliminação de resíduos da água de piscina por sulfato
de alumínio (forma polieletrólito na água) é feita após a floculação.
2.3. INTERAÇÃO ENTRE PARTÍCULAS COLOIDAIS
As interações entre partículas coloidais governam as propriedades dos
coloides e dependem da distância de separação e da quantidade de partículas
coloidais dispersas. As forças externas devidas ao campo da gravidade ou ao
cisalhamento também influenciam a interação e as colisões entre partículas.
Essas forças de interação entre as superfícies das partículas coloidais advêm
da natureza eletromagnética das interações entre a matéria.
Nas dispersões coloidais aquosas pode haver:
1) interação repulsiva de duplas camadas de cargas;

2) interação atrativa de van der Waals;
3) interação estérica repulsiva de cadeias de polímeros adsorvidos nas
partículas;
4) interação atrativa de polímeros;
5) interação de moléculas de solvente (solvatação)
6) interação hidrofóbica.
As partículas coloidais adquirem cargas elétricas na superfície, quando
expostas ao contato com solvente polar, por diferentes mecanismos, tais como:
dissociação de grupos da superfície e adsorção ou dissolução de íons da
superfície. Por isso o equilíbrio químico entre os prótons e a superfície de
óxidos é relevante para compreender o comportamento de dispersões
aquosas.
A carga da superfície da partícula influencia a distribuição dos íons da
solução na vizinhança, atraindo e repelindo contra íons e co-íons,
respectivamente. Essa distribuição de íons desde a superfície da partícula até
o interior da solução (meio de dispersão) gera diferentes potenciais e está
representada esquematicamente na Figura 1.
O potencial da interface entre a superfície da partícula e o interior da
solução do meio de dispersão diminui mais rapidamente à medida que
aumenta a força iônica, porque a dupla camada de cargas que se forma ao
redor da partícula é comprimida em direção à superfície pela concentração de
íons da solução. Portanto, as propriedades elétricas dos coloides são
governadas pelas interações repulsivas coulombianas.
No entanto, essa energia de repulsão entre as partículas não garante a
estabilidade das partículas dispersas. Por isso, na prática, dispersões coloidais
podem agregar-se e os agregados sedimentam-se rapidamente, como por
exemplo, no caso da dispersão de argila em água. As interações atrativas de
curto alcance de van der Waals induzem à agregação do sistema à medida que
as superfícies das partículas se aproximam umas das outras. Essas forças de
curto alcance são as mesmas provenientes da polarização e átomos e

moléculas (dipolos) constituintes dos sólidos dispersos no meio polar que
separa as partículas. Portanto, a energia total de interação (VT) é a soma
resultante das energias de repulsão (VR ) e de atração (VA) indicada na Figura
2. Esta é base da teoria DLVO, desenvolvida independentemente por Derjaguin
e Landau e Verwey e Overbeek, no final da década de 40, para explicar a
estabilidade cinética coloidal. A partir dos modelos e da formulação dessa
teoria, o estudo dos colóides passou a ser considerado com maior rigor
cientifico
Representação da dupla camada elétrica
3. SISTEMAS COLOIDAIS
3.1. GEL
Gel é um coloide no qual a interação do líquido com partículas muito
finas induz o aumento da viscosidade, tornando-se uma massa com partículas
organizadas no meio de dispersão formando uma rede de partículas
enfileiradas como um colar. Esses coloides formam uma rede com natureza
elástica e gelatinosa, tal como gelatina ou geleia de frutas, ou como um sólido

rígido como sílica gel, sendo muito usada em embalagens como agente
secante.
Exemplos de coloides
3.2. SOL
Sol é um colóide constituído de partículas sólidas, finamente divididas,
dispersas em um meio de dispersão líquido ou aéreo. Outras denominações -
hidrossol, organossol ou aerossol - são atribuídas segundo o meio de
dispersão utilizado: água, solvente orgânico ou ar, respectivamente.
Quanto à interação entre as moléculas da fase contínua e da fase
dispersa, os sóis são classificados em liofílicos, que apresentam partículas
dispersas com maior afinidade com o solvente, são mais estáveis e
semelhantes à solução verdadeira, e liofóbicos, cujas partículas não atraem
fortemente as moléculas de solvente e coagulam ou precipitam facilmente.
Essas dispersões coloidais mais concentradas formam sistemas mais viscosos
denominados pastas, utilizadas, por exemplo, na fabricação de creme dental.
A gelatina (meio contínuo: água, fase dispersa: proteínas) antes de
arrefecer é um exemplo de um sol (líquido). Porém quando arrefece
transforma-se num gel, onde a fase dispersa passa também a ser contínua.

Transição sol-gel e Efeito de Tyndall
3.3. ESPUMA
As espumas são sistemas coloidais onde um gás é borbulhado em um
meio de dispersão, onde bolhas de grande tamanho são formadas. O tipo de
espuma é definido de acordo com o meio de dispersão. Espumas sólidas
correspondem a gases dispersos em meios sólidos. Em Espumas líquidas a
dispersão é feita em meios líquidos.
Aplicações práticas da espuma

3.4. EMULSÃO
Emulsão são dispersões coloidais de um líquido em outro, geralmente
estabilizadas por um terceiro componente tensoativo (emulsificante) que se
localiza na interface entre as fases líquidas. Há dois tipos de emulsão,
conforme a proporção das fases: água em óleo, com gotículas de água
dispersas na fase contínua óleo, e óleo em água, gotículas de óleo dispersas
em água. O termo óleo refere-se à fase orgânica e água à fase aquosa. Esse
sistema coloidal é vastamente utilizado na apresentação de produtos
farmacêuticos (cremes), alimentícios (maionese, margarina, leite), industriais
(petróleo, lubrificantes, asfalto).
A maionese é uma emulsão estável apesar de ter óleo em sua
constituição e ovo (note que 65,5 % da composição do ovo é água). Isso ocorre
por causa dos chamados colóides protetores ou agentes emulsificantes ou
tensoativos, que no caso da maionese são as proteínas que se encontram na
gema do ovo. Essa película é hidrófila, o que significa que possui afinidade
com a água, assim a emulsão fica estabilizada e não ocorre como o exemplo
da água e óleo convencionais, em que, depois de pouco tempo, o óleo e a
água se separam, deixando de ser uma emulsão.

4. DUPLA CAMADA ELÉTRICA
Um dos principais fatores contribuintes está no fato das partículas
dispersas no meio dispersante possuírem cargas de mesmo sinal. Isso é
natural, visto que as partículas possuem a mesma composição, e assim sendo,
a mesma tendência a adquirirem cargas com sinais equivalentes. Esse
fenômeno ocorre ora pela ionização da própria partícula constituinte do coloide,
ora pela adsorção de íons.
Quando uma partícula acumula uma carga em sua superfície, está carga
tem como característica atrair outra de sinal oposto, gerando uma camada de
íons de carga oposta ao redor da partícula observada inicialmente. A presença
desses contra íons nas proximidades da superfície, em que uma das camadas
tem excesso de carga negativa, e a outra camada tem excesso de cargas
positivas, formam a dupla camada elétrica (DCE).
Como essas camadas possuem sinais opostos, ocorre uma repulsão
entre elas. E essa força de repulsão entre as partículas está fortemente
relacionada com a estabilidade de uma dispersão. Isso se deve ao fato, de que
nesses sistemas coloidais, haver uma força atrativa de Van der Waals. Em
sistemas em que essas forças atrativas são consideravelmente maiores, o
meio se torna instável, e as partículas sedimentam-se. Como há uma força de
repulsão proveniente da DCE, ocorre um equilíbrio, que é capaz de estabilizar
o sistema.
Esse fenômeno da estabilização do sistema está relacionado com as
principais funções de uma DCE: a influência que ela gera na adsorção por
atração eletrostática, além de afetar fortemente o estado de dispersão das
partículas. Um exemplo é na proteção das partículas coloidais através da
camada formada, tanto por repulsão entre cargas de mesmo sinal, quanto por
efeito estérico, evitando a aglutinação.
Evidentemente, a DCE, como um todo, é eletricamente neutra, e a sua
existência está condicionada a um excesso de carga na superfície mineral.
Prevalecendo essa neutralidade elétrica do sistema, a seguinte equação é
válida:

𝜎𝑜 = 𝜎𝑠 + 𝜎𝐺
em que:
𝜎𝑜: carga elétrica na superfície
𝜎𝑠 : carga elétrica na camada onde os íons permanecem fixos
𝜎𝐺: carga elétrica na camada difusa
Caso haja interesse, há maneiras de controlar a dupla camada
elétrica controlando o meio de suspensão. Isso pode ser feito através da
mudança dos íons dissolvidos, por meio da variação do pH e pela adição ou
remoção de surfactantes. Esses métodos mudam as interações entre as
partículas coloidais, podendo acabar com o equilíbrio dado no sistema.
Formação da dupla camada elétrica
Há vários exemplos práticos de soluções em que a presença da DCE é
indispensável. Aparecem nas tintas, no sangue e no leite, por exemplo. Nesse
último, a DCE é responsável por manter a homogeneidade do leite, já que a
sua gordura se mantém estabilizada graças ao efeito estabilizante da dupla
camada elétrica, em que há uma força de repulsão (entre camadas de mesmo
sinal) e uma força atrativa (força de Van der Waals), que se equilibram.

5. ELETROCAPILARIDADE E EQUAÇÃO DE LIPPMAN
Para o entendimento das interações e da composição das interfaces
eletrodo-solução, foi desenvolvido diversos modelos, com seu constante
aperfeiçoamento, para compreender os processos ocorridos nas técnicas
voltamétricas. Com o encontro das duas fases nesta interface é comum ocorrer
um rearranjo de espécies que leva a um equilíbrio entre as fases, o que gera
um potencial no sistema e nesta região interfacial. A constante de
proporcionalidade entre o potencial aplicado e a carga devida às espécies
ordenadas na região interfacial na solução é a capacitância desta camada. Tal
constante consegue ser medida por métodos de impedância, porém
antigamente era medida pela eletrocapilaridade, o que permitiu a criação dos
primeiros modelos teóricos sobre a capacitância em função do potencial,
usando o eletrodo de mercúrio.
A eletrocapilaridade trata-se de um fenômeno no qual com o uso do
campo elétrico é possível realizar a variação de tensão superficial de um
líquido. Por curiosidade, a eletrocapilaridade possui aplicações recentes, como
a aplicação desta técnica em dispositivos destinados à determinação da
quantidade de glicose presente em fluidos fisiológicos. Com o crescimento dos
afetados pela Diabetes e pelo interesse em sistemas miniaturizados de
análises biológicas, a aplicação se mostrou viável e com vantagens em relação
aos métodos anteriores. A capacidade de manipular microgotas faz a
eletrocapilaridade também ser utilizada na construção de equipamentos
eletrônicos como microchaves, nas quais gotas de mercúrio são movimentadas
por campos eletrostáticos.
Segundo o modelo de Helmholtz para descrever o comportamento da
interface eletrodo-solução, as cargas positivas e negativas estavam ordenadas
nos dois lados da interface o que torna comparável a um capacitor de placas
paralelas. A equação de Lippmann permite o cálculo do valor da carga
superficial existente numa dupla camada elétrica, relacionando com a tensão
superficial e com o potencial. Assim, neste caso que se assemelha com o
capacitor de placas paralelas, temos uma equação que rege o fenômeno da
eletrocapilaridade. Temos que a variação da tensão superficial é igual à soma

do trabalho elétrico do deslocamento das cargas com o somatório dos
potenciais químicos multiplicados por constantes das substâncias.
𝑑𝛾 = −𝑞𝑀 ∗ 𝑑𝜑 − ∑ Г𝑖
𝑖
𝑑𝜇𝑖
Considerando um sistema de uma gota de mercúrio em uma solução
não-iônica qualquer, não há variação da concentração das espécies
dissolvidas, assim a variação da energia livre de Gibbs é nula. Sendo a
diferença de potencial entre os eletrodos a única grandeza medida, temos:
𝑑𝛾 = −𝑞𝑀 ∗ 𝑑𝜑1
𝑑𝛾 = −𝑞𝑀 ∗ 𝑑𝜀 − 𝑞𝑀 ∗ 𝑑𝜑𝑅
Não havendo variação no valor do potencial de referência,
chegamos a equação de Lippmann que permite o cálculo em qualquer ponto da
curva de eletrocapilaridade.
(𝑑𝛾
𝑑𝜀)
𝑥𝑖
= −𝑞𝑀
Como o capacitor de placas paralelas pode ser associado a este caso de
interação eletrodo-solução, é possível escrever, analogamente, para a dupla
camada uma equação que rege seu comportamento. Sendo a equação do
capacitor:
𝐶 = 𝑑𝑞
𝑑𝑉
Então, para a dupla camada:
𝐶 = 𝑑𝑞𝑀
𝑑𝜑1=
𝑑𝑞𝑀
𝑑𝜀= − (
𝑑2𝛾
𝑑𝜀2)
𝑥𝑖
Sendo C a capacitância da dupla camada elétrica.
É importante salientar que a associação da DCE com um capacitor é um
modelo muito efetivo para soluções suficientemente concentradas, isto é,
soluções com pouca carga dispersa, mas sim comprimidas dentro do plano de
Helmholtz-Perrin. Quando a solução está diluída, é necessário usar os modelos

de Gouy-Chapman, onde a carga dispersa na solução sofre influência tanto das
forças elétricas, quanto das térmicas. Outros modelos foram propostos ao
decorrer do tempo, como Grahame em 1947 e Bockris e Muller com outro mais
preciso.
O modelo de Gouy-Chapman leva em conta o potencial e a dependência
do potencial com a concentração, variando a capacitância diferencial. Sendo a
camada menos compacta que no modelo anterior e sim com certa espessura
que permite a movimentação de elétrons nela. Em 1924, Stern juntou os dois
modelados apresentados e o modelo misto pode ter sua capacitância calculada
pela equação de Lippmann:
∂ γ / ∂∆ φ = - q M = q S
Sendo γ a tensão superficial, ∆φ as mudanças de potencial na interface
e θ a carga no metal (M) e na solução (S).
Utilizando as considerações do capacitor de placas paralelas e as novas
ideias de Gouy-Chapman chegamos que a capacitância diferencial é
relacionada com a capacitância de Helmhotlz-Perrin (HM) e com a capacitância
de Gouy-Chapman(GC). Desse modo:
(1/ Cd) = (1/CHM) + (1/CGC)
6. MODELOS DE DUPLA CAMADA
6.1. MODELO DE HELMHOLTZ (1853)
Conceitualmente, quando um metal se dissolve de forma contínua, ele
tende a ficar cada vez mais carregado negativamente, devido à passagem de
íons positivos contínuos na solução. Assim, há um excesso de cargas
negativas na superfície do metal, e um excesso de cargas positivas na solução,
ou seja, uma separação de cargas. Como há uma interação entre as cargas, é
criado um campo elétrico. Essas cargas estão separadas em duas camadas,
explicando o nome de “dupla camada”.
A separação das cargas nessa dupla camada é explicada pelo modelo
de DCE mais simples, proposto por Helmholtz. Nesse modelo, acreditava-se

que os íons mantinham-se rigidamente fixos nas proximidades da superfície do
sólido, separados por uma fina película de moléculas de água, formando uma
espécie de condensador de placas paralelas em um capacitor.
Resumidamente, o modelo consiste em duas camadas. Internamente, a
camada interna de Helmholtz. Essa camada é compreendida por moléculas de
água absorvidas e alguns ânions adsorvidos. Além desta, há também uma
camada externa de Helmholtz, que é compreendida por cátions hidratados. Ao
todo, essa camada apresenta uma espessura de aproximadamente 1 nm.
Para essa situação, foi considerado que o potencial das cargas variaria
linearmente entre as camadas. Entretanto, para sistemas reais, essa variação
de potencial não é constante e muda de acordo com a concentração de íons
em solução, além do potencial que é aplicado na superfície do eletrodo. Um
dos grandes erros desse modelo foi em não considerar o movimento
Browniano dos contra íons, ignorando a existência da camada difusa.
Modelo da DCE proposto por Helmholtz
No eletrodo negativo, o potencial 𝑈 = 𝛷𝑚𝑒𝑡𝑎𝑙, enquanto no eletrólito o
potencial 𝑈 = 𝛷𝑠𝑜𝑙𝑢çã𝑜. Garantindo a diferença de potencial, tem-se:
(𝛷𝑚𝑒𝑡𝑎𝑙 - 𝛷𝑠𝑜𝑙𝑢çã𝑜) = 𝑄/𝐶.

6.2. MODELO DE GOUY – CHAPMAN (1910 – 1913)
O modelo de Helmholtz foi modificado por Gouy (1910) e Chapman
(1913). Neste, a dupla camada seria uma superfície rígida e carregada,
neutralizada por uma nuvem de íons com cargas opostas. Gouy e Chapman
conceberam a existência de uma camada difusa de contra íons movimentando-
se sob a influência da energia térmica. Neste modelo, a concentração do
eletrólito é levada em consideração, em que a maior quantidade de espécies
estaria imediatamente ao lado do eletrodo, de forma que a atração eletrostática
é superior aos efeitos térmicos. Dessa forma, a concentração de espécies é
afetada por dois fatores: distância do eletrodo (camada de carga difusa) e
carga aplicada ao eletrodo.
No entanto, o modelo considerava os íons como cargas puntiformes,
acarretando em discrepâncias entre a teoria e os resultados experimentais,
visto que a espessura da camada dupla considerada era maior que a
calculada. Isso limitava a aplicabilidade do modelo às superfícies planas com
baixa densidade de carga ou para grandes distâncias de afastamento da
superfície sólida.
Modelo de Gouy-Chapman
Vale ressaltar que nesse modelo, a distribuição dos íons na camada
difusa obedece à lei de Fick, e a dependência da energia térmica na
distribuição desses íons obedece a lei de distribuição de energia de Boltzmann
e a Lei eletrostática de Poisson.

6.3. MODELO DE STERN (1924)
O modelo proposto por Stern é uma união dos dois modelos descritos
anteriormente, onde há a existência de uma camada compacta de íons nas
proximidades do eletrodo (modelo de Helmholtz), seguida por uma camada
difusa de íons (modelo de Gouy-Chapman), aproximando o tratamento
matemático para a existência de dois capacitores ligados em série, que
representam as camadas compacta e difusa. Além dessas duas camadas,
Stern propôs que alguns íons eram adsorvidos pela superfície no plano d,
formando uma camada conhecida como Camada Stern.
Além disso, nesse modelo, os íons têm tamanho finito. Assim sendo, não
podem se aproximar da superfície mais perto do que alguns nm. Dessa forma,
os primeiros íons da dupla camada difusa não estão na superfície, mas à
alguma distância d da mesma, que é tomada como o raio do íon. Como
resultado, o potencial e concentração da parte difusa da camada é baixo o
suficiente para justificar o tratamento dos íons como cargas pontuais.
Modelo de Stern
Por se tratar de uma mistura de dois modelos em que o cálculo da
variação de potencial era calculado de forma divergente, o modelo de Stern
estabeleceu duas regiões para a variação de potencial: a) Uma linear:
relacionada aos contra íons que permanecem fixos nas proximidades da
superfície sólida (modelo de Helmholtz) e b) Outra exponencial: corresponde

aos íons que se movimentam na camada difusa sob a influência da energia
térmica (modelo de Gouy-Chapman)
Variação de potencial para os modelos de Helmholtz e Gouy-Chapman
6.4. MODELO DE GRAHAME (1947)
Apesar de Stern distinguir os íons adsorvidos no eletrodo e os da dupla
camada, foi Grahame que desenvolveu o modelo que é constituído por três
regiões:
Modelo de Grahame
a) Região dos íons especificamente adsorvidos (íons mais próximos ao
eletrodo – IHP). Aqui, há um plano de máxima aproximação com adsorção de
moléculas de solventes e, possivelmente, de ânions especificamente
adsorvidos, em que o potencial varia linearmente com a distância.
b) Região dos íons solvatados e não-especificamente adsorvidos (OHP).
Apresenta espécies hidratadas (normalmente cátions), em que o potencial varia
linearmente com a distância.

c) Região difusa que está fora do OHP. Possui excesso de cátions ou ânions,
em que o potencial varia exponencialmente com a distância.
Comparação entre os modelos de Helmholtz, Gouy-Chapman e Stern,
respectivamente
7. ESTABILIDADE E COAGULAÇÃO DE DISPERSÕES COLOIDAIS
7.1. ESTABILIDADE
A estabilidade dos coloides depende primordialmente das propriedades
da fase dispersa, que como foi mencionado em 2.2 podem ter afinidades do
tipo liofílica (afinidade fraca) ou liofóbica (afinidade forte), onde o termo lio
refere-se ao meio dispersante.
Em geral, os coloide liofílicos são bastante fáceis de preparar, bastante
estáveis e razoavelmente simples de reconstruir. Os coloides liofóbicos
geralmente são menos estáveis e são excepcionalmente difíceis de reconstruir.
Um exemplo de sistema liofílico é o sabão disperso na água. Já como exemplo
de sistema liofóbico tem o óleo suspenso na água, pelo uso de uma técnica de
dispersão por ultrassons.
A rigidez inerente dos coloides não fluídos, tais como as espumas
sólidas ou os sóis sólidos é, em geral, o fator principal que determina a sua
estabilidade. Muitos coloides líquidos são estabilizados pela a adição de
surfactantes (também chamados anfifílicos), os quais são moléculas que têm
uma região liofílica e uma liofóbica.

7.1.1. MECANISMO
O mecanismo da estabilização baseia-se na formação de micelas,
podendo ser normais ou invertidas.
Micelas
Nas micelas normais as moléculas do surfactante envolvem a substância
hidrofóbica (óleo, por exemplo). Esta é uma forma bastante eficiente de
estabilizar uma emulsão de óleo na água, pois o surfactante cria uma barreira
mecânica que envolve cada gotícula de óleo, impedindo que estas se juntem
quando chocam entre si. A existência de cargas do mesmo sinal, associadas
às cabeças hidrofílicas, é um fator adicional de estabilidade devido às
repulsões eletrostáticas entre as micelas.
Micela Invertida
Nas micelas invertidas as moléculas do surfactante envolvem a
substância hidrofílica (água, por exemplo). Neste caso as cabeças hidrofílicas
ficam em contato com a água e as caudas hidrofóbicas ficam em contato com o

óleo, blindando as gotículas de água e impedindo desta forma que estas se
juntem quando chocam entre si.
É graças à formação das micelas que os sabões e detergentes (que não
são os únicos surfactantes) dispersam a gordura na água. Por exemplo, a
estabilidade do leite deve-se à caseína (proteína) e a da maionese à lecitina
(proteína) presente na gema de ovo, onde a caseína e a lecitina funcionam
como surfactantes. Por outro lado, a adição das enzimas presentes no coalho
ao leite destrói as micelas de caseína, o que permite que estas se aglutinem e
deem origem ao queijo depois da extração do soro.
Os surfactantes têm também um papel importante na estabilização das
espumas líquidas, pois as moléculas do surfactante que estão à superfície
fazem diminuir a tensão superficial da água. A água pode assim distribuir-se
por películas finas, ao invés de procurar concentrar-se num volume o mais
compacto possível de forma a minimizar a área de contato com o ar. É por
essa razão que as bolas de sabão são tão estáveis.
A proporção de água, óleo e surfactante deve ser controlada, para que
não forme estruturas muito mais complexas do que as micelas. Caso essas
sejam formadas, alterariam fortemente fatores da substância como sua
estabilidade, por exemplo.
Provavelmente a situação mais comum que contribui para a estabilidade
coloidal é o fato de as partículas dispersas adquirirem cargas do mesmo sinal
(visto que têm a mesma composição, não é de surpreender que tendem a
acumular o mesmo tipo de carga). Este fenômeno deve-se tanto à adsorção de
íons do meio de dispersão, ou de moléculas de surfactante carregadas, quanto
à ionização de algumas moléculas ou partes de moléculas situadas à superfície
das partículas dispersas.
Por exemplo, se a partícula coloidal acumular uma carga negativa à
superfície, esta carga vai atrair os íons de carga positiva do meio de dispersão
criando-se uma atmosfera difusa de íons de carga contrária (neste caso
positiva) à volta da partícula coloidal, o que dá origem à criação de uma dupla
camada elétrica, também já falada no item 4. É esta dupla camada elétrica que
"protege" as partículas coloidais, quando estas chocam, pois as atmosferas das
partículas coloidais têm carga do mesmo sinal e consequentemente repelem-

se, além de funcionarem como barreiras físicas que evitam a aglutinação das
partículas coloidais.
A força de repulsão elétrica não é, porém, a única força em jogo: existe
também a força atrativa de Van der Waals, sendo o balanço destas duas forças
que dita a estabilidade, ou a falta dela, de um coloide:
Balanço entre as forças de Van der Waals e repulsão elétrica
Esta teoria foi desenvolvida no começo da década de 40 por dois grupos
de cientistas, Boris Derjagin e Lev Landau na Rússia, e Evert Verwey e Theo
Overbeek na Holanda. Ambos os grupos publicaram as suas ideias após a
Segunda Guerra Mundial e a teoria ficou democraticamente conhecida por
teoria de Derjagin-Landau-Verwey-Overbeek, universalmente abreviada para
"DLVO".
A dupla camada elétrica pode ser controlada modificando o meio de
suspensão, quer pela variação do pH, quer pela mudança dos íons dissolvidos,
quer pela a adição de surfactantes, quer pela a adição de enzimas que
destroem os surfactantes. É assim possível manipular as interações entre
partículas coloidais, podendo-se, inclusive, fazê-las passar de
predominantemente repulsivas para predominantemente atrativas, e vice-versa.
7.2. COAGULAÇÃO
A coagulação é empregada, principalmente, para a remoção de material
em suspensão ou coloidal. As partículas coloidais não sedimentam e não
podem ser removidas por processos de tratamento físicos convencionais.
Os coloides possuem propriedades elétricas que criam uma força de
repulsão que impede a aglomeração e a sedimentação. Se suas características

não forem alteradas, permanecem no meio líquido. Para que as impurezas
possam ser removidas, é preciso então alterar algumas características por
meio da coagulação, floculação, sedimentação (ou flotação) e filtração.
As condições de mistura e de floculação para a agregação de coloides
orgânicos serão diferentes daquelas empregadas para coloides inorgânicos: as
superfícies hidrofílicas destas partículas orgânicas reagem diferentemente à
adição de coagulante.
Em geral, coagulante é o produto químico que é adicionado para
desestabilizar as partículas coloidais de modo que possa formar o floco. Em
geral são utilizados sais de alumínio e ferro. Floculante é o produto químico,
geralmente orgânico, adicionado para acentuar o processo de floculação. A
maioria dos floculantes usados consiste em polieletrólitos tais como derivados
de poliacrilamida e de poliestireno.
A coagulação é o processo de desestabilização das partículas coloidais
de modo que o crescimento da partícula possa ocorrer em consequência das
colisões entre partículas. O papel do coagulante aqui é desestabilizar a
suspensão coloidal reduzindo todas as forças atrativas, desse modo abaixando
a barreira de energia e permitindo partículas de se agregarem.
Dependendo das propriedades físicas e químicas da solução, do
poluente e do coagulante, um número de mecanismos de coagulação (por
exemplo, neutralização da carga, compressão da dupla camada, formação de
pontes e arraste) foram postulados. A dosagem do produto químico entrega o
coagulante como um sal que se dissocia na solução com hidrólise do cátion de
alumínio (e de ânions associados) que determinam a especiação da solução e
pH. Por exemplo, adição de alumínio (isto é, sulfato de alumínio) acidifica a
água.
A coagulação resulta de dois fenômenos: o primeiro, essencialmente
químico, consiste nas reações do coagulante com a água e na formação de
espécies hidrolisadas com carga positiva e depende da concentração do metal
e pH final da mistura; o segundo, fundamentalmente físico, consiste no
transporte das espécies hidrolisadas para que haja contato entre as impurezas
presentes na água. O processo é muito rápido, variando desde décimos de
segundo a cerca de 100 segundos, dependendo das demais características
(pH, temperatura, condutividade elétrica, concentração de impurezas, etc.). Daí

em diante há necessidade de agitação lenta, para que ocorram choques entre
as impurezas, que se aglomeram formando partículas maiores, denominadas
flocos, que podem ser removidos por sedimentação, flotação ou filtração
rápida. Essa etapa é denominada floculação. Uma característica essencial da
floculação da água residual é a eliminação dos sólidos em suspensão e do
material orgânico tanto quanto possível.
O processo de coagulação/floculação pode ser usado como pré-
tratamento antecedente ao tratamento biológico a fim de aumentar a
biodegradabilidade da água residual durante o tratamento biológico. Se o
tratamento físico-químico feito no estágio inicial do tratamento de água
residuária for eficaz, a carga orgânica em toda a fase biológica subsequente ao
tratamento estará então reduzida consideravelmente. A planta de tratamento
completa será mais compacta e terá maior eficiência de energia.
As dosagens dos coagulantes variam em grande escala para maximizar
a eficiência de remoção dos poluentes, usando doses mínimas no pH ótimo. As
condições ótimas para a coagulação podem ser determinadas fazendo-se o Jar
Test. Estes testes podem ser usados para estabelecer o melhor tipo e a melhor
concentração do coagulante, as condições apropriadas de mistura e as taxas
de sedimentação floculenta para melhor remoção orgânica e/ou da toxicidade.
7.2.1. MECANISMOS
A coagulação resulta de dois mecanismos básicos: coagulação
pericinética e eletrocinética, na qual o potencial zeta é reduzido por íons ou
colóides de carga contrária a um nível abaixo das forças atrativas de van der
Waals, e coagulação ortocinética, na qual as micelas se agregam e formam
blocos que aglomeram as partículas coloidais. A adição de cátions com altas
valências abaixa a carga da partícula e a distância efetiva da dupla camada,
desse modo reduzindo o potencial zeta.
Porém, atualmente considera-se a coagulação como o resultado
individual ou combinado da ação de quatro mecanismos distintos:
7.2.1.1. COMPREENSÃO DA DUPLA CAMADA ELÉTRICA
A introdução de um eletrólito indiferente, ou seja, o qual não tem
característica de hidrólise ou de adsorção (como sais simples, por exemplo,

cloreto de sódio), em um sistema coloidal causará aumento na densidade de
cargas na camada difusa e diminuirá a “esfera” de influência das partículas,
ocorrendo a coagulação por compressão da camada difusa. Concentrações
elevadas de íons positivos e negativos (força iônica grande) na água acarretam
acréscimo do número de íons na camada difusa, que, para se manter
eletricamente neutra, necessariamente tem seu volume reduzido (diminuição
da espessura), de modo tal que as forças de Van der Waals sejam dominantes,
eliminando a estabilização eletrostática.
Dois aspectos devem ser destacados neste mecanismo de coagulação:
a) a quantidade de eletrólitos para conseguir a coagulação é, praticamente,
independente da concentração de coloides na água;
b) para qualquer quantidade adicionada de eletrólitos é impossível estabilizar
novamente as partículas coloidais, ou seja, a reversão de carga das mesmas,
que passa a ser positiva.
7.2.1.2. ADSORÇÃO E NEUTRALIZAÇÃO
A desestabilização de uma dispersão coloidal consiste nas interações
entre coagulante-coloide, coagulante-solvente e coloide-solvente.
Como o coagulante se dissolve, os cátions servem para neutralizar a
carga negativa do coloide. Isso ocorre antes da formação visível do floco, e a
agitação rápida é efetiva nesta fase. Micro flocos são então formados os quais
retém a carga positiva na faixa ácida devido à adsorção de H+. Esses micro
flocos também servem para neutralizar e cobrir as partículas coloidais.
No caso de espécies hidrolisadas de alumínio ou de ferro ou de
polímeros sintéticos catiônicos, é comum ocorrer adsorção específica, causada
pela interação entre coagulante e coloide.
O mecanismo de adsorção-neutralização de carga é muito importante
quando o tratamento é realizado através da tecnologia de filtração direta, pois
as partículas desestabilizadas são retidas no meio granular dos filtros.
7.2.1.3. VARREDURA
Dependendo da quantidade adicionada de coagulante, do pH da mistura
e da concentração de alguns tipos de íons na água, poderá ocorrer a formação
de precipitados do tipo Al(OH)3 ou Fe(OH)3. Em geral, os flocos obtidos com

esse mecanismo são maiores e sedimentam ou flotam mais facilmente que os
flocos obtidos com a coagulação realizada no mecanismo de adsorção e
neutralização de cargas. A floculação aglomera os coloides com o floco
hidratado do óxido. Nesta fase, a superfície de adsorção é ativa. Coloides não
inicialmente adsorvidos são removidos por emaranhamento no floco. Os
mecanismos de coagulação por adsorção e neutralização de cargas e por
varredura, quando é utilizado sulfato de alumínio, podem ocorrer segundo os
caminhos indicados abaixo:
Caminhos para a coagulação por adsorção-neutralização de carga e por
varredura utilizando sulfato de alumínio.
7.2.1.4. ADSORÇÃO E FORMAÇÃO DE PONTES
O mecanismo de adsorção e formação de pontes (interparticle bridging)
caracteriza-se por envolver o uso de polímeros de grandes cadeias
moleculares (massa molar > 1000000), os quais servem de ponte entre a
superfície à qual estão aderidos e outras partículas.

8. REFERÊNCIAS
BETTEGA, Marcio H. F.. Introdução à Teoria Quântica do Espalhamento:
do Espalhamento por um Potencial ao Problema de Muitos
Corpos. Disponível em: <http://fisica.ufpr.br/bettega/XIII-EBEE/aula1.pdf>.
Acesso em: 04 set. 2018.
DOUBLE LAYER THEORIES. Disponível em:
<https://www.garmanage.com/atelier/index.cgi?path=public&B&Energy_storage
&B&Supercapacitors&B&Double_layer&&id=psyitefg>. Acesso em: 13 set.
2018.
ELETRIC DOUBLE LAYER. Disponível em:
<http://web.nmsu.edu/~snsm/classes/chem435/Lab14/double_layer.html>.
Acesso em: 11 set. 2018.
FERNANDES, Julio Cesar Bastos; KUBOTA, Lauro Tatsuo; OLIVEIRA NETO,
Gracilliano de. ELETRODOS ÍON-SELETIVOS: HISTÓRICO, MECANISMO
DE RESPOSTA, SELETIVIDADE E REVISÃO DOS CONCEITOS. Quimica
Nova, Campinas, v. 24, n. 1, p.120-130, maio 2001. Disponível em:
<http://www.scielo.br/pdf/%0D/qn/v24n1/4459.pdf>. Acesso em: 18 set. 2018.
FIGUEREDO, R.C.R.; RIBEIRO, F.A.L. e SABADINI, E. Ciência de espumas -
aplicação na extinção de incêndios. Quimica Nova, v. 22, n. 1, p. 126-130,
1999
FÍSICA, À Luz da. Controlo dos Coloides. Disponível em:
<http://cftc.cii.fc.ul.pt/PRISMA/capitulos/capitulo3/modulo6/topico4.php>.
Acesso em: 10 set. 2018.
HUIBERS, P. http://www. surfactants.net, Instituto de Tecnologia de
Massachusetts, Departamento de Química, maio 1999.
HUSSAINI. Understanding Corrosion. Disponível em:
<http://faculty.kfupm.edu.sa/ME/hussaini/Corrosion%20Engineering/02.05.04.ht
m>. Acesso em: 07 set. 2018.
JUNIOR, Miguel Jafelicci; VARANDA, Laudemir Carlos. Química Nova na
Escola: O mundo dos colóides. 9. 1999. Disponível em:

<http://qnesc.sbq.org.br/online/qnesc09/quimsoc.pdf>. Acesso em: 22 ago.
2018.
MAXWELL; Coagulação Química - PUC - Rio, Capítulo 4 p. 40-57
NECKEL, Itamar Tomio. CRESCIMENTO E MORFOLOGIA DE LIGAS DE
CoXFe100-X ELETRODEPOSITADAS SOBRE Si(111) TIPO-n. 2009. 108 f.
Dissertação (Mestrado) - Curso de Engenharia e Ciência de Materiais,
Universidade Federal do Paraná, Curitiba, 2009.
NOÇÕES BÁSICAS DE ELETROQÚIMICA: USP. Disponível em:
<https://edisciplinas.usp.br/pluginfile.php/3083350/mod_resource/content/1/Con
ceitos%20de%20eletroquimica.pdf>. Acesso em: 13 set. 2018.
RANGEL, Rita de Cássia Cipriano. Aplicação da eletrocapilaridade na
manipulação de microgotas. 2008. 87 f. Dissertação (Mestrado) - Curso de
Engenharia, Universidade Estadual Paulista, Sorocaba, 2008. Disponível em:
<https://repositorio.unesp.br/bitstream/handle/11449/88367/rangel_rcc_me_bau
ru.pdf?sequence=1>. Acesso em: 18 set. 2018.
SCHONS, Elenice. Fenômenos Interfaciais; Catalão; CETM, 2013. Color.
SCHONS, Elinece. Tipos e caracterização da interface. Disponível em:
<https://cetm_engminas.catalao.ufg.br/up/596/o/fen_int_1.pdf>. Acesso em: 07
set. 2018.
VIDOTTI, Marcio. Eletroquímica 6. Disponível em:
<http://www.quimica.ufpr.br/mvidotti/cq049-aula06.pdf>. Acesso em: 11 set.
2018.
VIDOTTI, Marcio. Eletroquímica 7. Disponível em:
<http://www.quimica.ufpr.br/mvidotti/cq049-aula07.pdf>. Acesso em: 11 set.
2018.
VINCENT, B., Departamento de Química da Universidade de Bristol, Colóides
e superfícies, http:// www.bristol.ac.uk/Depts/Chemistry/ mscsurf.htm, maio
1999.
AZEVEDO, Antonio Carlos de; BONUMÁ, Angélica Silveira. Partículas
coloidais, dispersão e agregação em latossolos. Disponível em:
<http://www.redalyc.org/html/331/33134246/>. Acesso em: 05 set. 2018.


















