
FENÔMENOS DE SUPERFÍCIE E...
22
FENÔMENOS DE SUPERFÍCIE E ELETROQUÍMICA ISOTERMAS DE ADSORÇÃO DE LANGMUIR, BET E GIBBS GRUPO I: Amanda Ferreira Nº USP: 9442177 Giulia Fantoni Carvalho Castro Nº USP: 9359921 Mariah Lopes de Toledo Nº USP: 7672761 Otávio Augusto da Silva Nº USP: 7672903 Thiago Luis Dorigan da Silva Nº USP 9270871 Vinícius Marcondes Nº USP: 8077327 Prof.ª. Marivone Nunho Sousa Lorena Agosto/2018
Transcript of FENÔMENOS DE SUPERFÍCIE E...

FENÔMENOS DE SUPERFÍCIE E ELETROQUÍMICA
ISOTERMAS DE ADSORÇÃO DE LANGMUIR, BET E GIBBS
GRUPO I:
Amanda Ferreira Nº USP: 9442177
Giulia Fantoni Carvalho Castro Nº USP: 9359921
Mariah Lopes de Toledo Nº USP: 7672761
Otávio Augusto da Silva Nº USP: 7672903
Thiago Luis Dorigan da Silva Nº USP 9270871
Vinícius Marcondes Nº USP: 8077327
Prof.ª. Marivone Nunho Sousa
Lorena
Agosto/2018

2
ÍNDICE
1. INTRODUÇÃO 3
2. ISOTERMAS DE ADSORÇÃO 5
2.1 Definição 5
2.2 Tipos 5
2.3 Modelo de Langmuir 6
2.4 Teoria das Multicamadas de BET (Brunauer, Emmet e Teller) 9
2.5 Isoterma de adsorção de Gibbs 11
3. APLICAÇÕES 16
3.1 Adsorção de cádmio e chumbo em diferentes classes de solos brasileiros – Aplicação dos
modelos de Langmuir e Freundlich
3.2. Área Específica de catalisadores 17
REFERÊNCIAS 20

3
1. INTRODUÇÃO
Adsorção é o processo por meio do qual constituintes de um fluido são retidos sobre
um sólido. Neste processo de acumulação e concentração seletiva, a substância que tem
constituintes retidos é chamada de adsorvato e o sólido que adsorve é chamado de adsorvente
ou substrato.
A adsorção é um processo que depende da velocidade com que a substância se difunde
através da solução, em direção ao sólido e, portanto, não é instantânea. Esta velocidade pode
ser determinada observando as mudanças do grau de recobrimento com o tempo. O processo
inverso da adsorção é a dessorção.
Grau (ou fração) de recobrimento é quão coberta está uma superfície. Ele normalmente
é expresso da seguinte forma:
𝜃 =𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑠í𝑡𝑖𝑜𝑠 𝑑𝑒 𝑎𝑑𝑠𝑜𝑟çã𝑜 𝑜𝑐𝑢𝑝𝑎𝑑𝑜𝑠
𝑛ú𝑚𝑒𝑟𝑜 𝑑𝑒 𝑠í𝑡𝑖𝑜𝑠 𝑑𝑒 𝑎𝑑𝑠𝑜𝑟çã𝑜 𝑑𝑖𝑠𝑝𝑜𝑛í𝑣𝑒𝑖𝑠
Podemos, também, expressar o grau de recobrimento em função do volume adsorvido:
θ = V/V∞, onde V∞ é o volume necessário para o adsorvato recobrir todo o sólido.
A adsorção possui variação da energia livre de Gibbs negativa, sendo, portanto, um
processo espontâneo. No entanto, as moléculas do líquido adsorvido ficam limitadas ao sólido
e, por isso existe uma redução do grau de liberdade do líquido, acarretando uma variação de
entropia negativa. Da fórmula ∆H = ∆G - T∆S podemos perceber que a adsorção é um
processo exotérmico.
A adsorção pode ocorrer de duas maneiras: o líquido e o sólido podem formar ligações
químicas, o que é chamado de quimissorção; ou o adsorvato pode ficar retido apenas por
interações intermoleculares, caso chamado de fisissorção.
Apesar de fracas, as interações envolvidas na fisissorção (adsorção física) são de longo
alcance, possibilitando a formação de múltiplas camadas moleculares sobrepostas, coisa que
não ocorre na adsorção química (quimissorção), que tem a formação de apenas uma camada
adsorvida normalmente por ligações covalentes.
A velocidade das quatro etapas abaixo define a cinética da adsorção:
1) O movimento do fluido que será adsorvido através da solução até a interface com
o sólido (camada limite).
2) Transporte do adsorvato da camada limite até a entrada dos poros, por difusão
(difusão externa).

4
3) Transporte do adsorvato através dos poros da partícula por uma combinação de
difusão molecular através do líquido contido no interior dos poros e difusão ao
longo da superfície do adsorvente (difusão interna).
4) Adsorção, ligação do adsorvato em um sítio disponível do adsorvente, envolvendo
vários mecanismos, tais como: adsorção física, adsorção química, troca iônica,
precipitação, complexação.
Figura 1: Quatro etapas da adsorção.
Vale muito a pena relembrar que o processo de adsorção pode ser classificado como
um processo físico ou químico, dependendo do tipo de força envolvido, já que isso
influencia muito nas caracterizações químicas à base de adsorção. Assim, para reforçar
destaca-se que o primeiro processo pode ser comparado com a condensação de vapor para
formação de um líquido, que só é importante a temperaturas abaixo da temperatura crítica
do gás. A adsorção química ou quimissorção envolve interações específicas entre o
adsorvente e o adsorvato com energia quase tão alta quanto a de formação de ligações
químicas.
A Tabela 1 abaixo mostra as principais diferenças entre os dois tipos de adsorção.

5
Tabela 1 - Principais diferenças entre os dois tipos de adsorção.
2. ISOTERMAS DE ADSORÇÃO
2.1 Definição
O gás desprendido e o adsorvido encontram-se em equilíbrio dinâmico e o nível de
recobrimento do campo externo do material necessita da pressão do gás em equilíbrio. A
variação 𝜃 com a pressão, a uma temperatura estática constitui em uma isoterma de adsorção.
Estas constituem sinuosidades, as quais indicam a forma como o adsorvente
verdadeiramente adsorverá aos constituintes e se a apuração requerida consegue ser atingida.
Essas curvas fornecem uma aproximação da porção máxima de soluto que será adsorvida
além de dados que determinam se o adsorvente será economicamente realizável para a
purificação de um líquido.
2.2 Tipos
As principais classificações de isotermas de adsorção estão na figura abaixo, na qual qe
representa a porção extremo de soluto retida no adsorvente e Ce é a concentração de
equilíbrio.

6
Figura 2: Tipos de isotermas de adsorção.
A isoterma linear é constituída de uma reta a qual se inicia na origem e sugere que a
quantidade adsorvida é equivalente a concentração do fluído, não correspondendo à
capacidade máxima de adsorção. As isotermas côncavas são aquelas simpatizantes, por
excisar quantidade elevadas mesmo em baixos níveis de concentração de adsorvato do fluído.
Isotermas convexas são denominadas desfavoráveis, pois possuem baixa capacidade de
remoção em baixas concentrações. Essas são escassas, contudo muito relevantes para
compreender o processo de regeneração da transferência de massa do sólido para a fase fluida.
Agora, quando o estudo do fenômeno de adsorção é feito com o objetivo de se obter
informações sobre a área específica e a estrutura porosa de um sólido, a construção de uma
isoterma de adsorção é de fundamental importância, pois sua forma revela muitos detalhes
sobre as características do material. A isoterma mostra a relação entre a quantidade molar de
gás n adsorvida ou dessorvida por um sólido, a uma temperatura constante, em função da
pressão do gás.
Abaixo, são destacadas na Figura 3 os tipos de isotermas de adsorção obtidas após
análises experimentais de adsorção.
O formato da isoterma é função do tipo de porosidade do sólido. Várias são as formas
de isotermas conhecidas até hoje, porém, todas são variações de seis tipos principais. Os cinco
primeiros tipos foram primeiramente sugeridos por Brunauer em 1938, sendo o sexto tipo
proposto mais tarde.
A isoterma do tipo I é característica de sólidos com microporosidade. As isotermas do
tipo II e IV são típicas de sólidos não porosos e de sólidos com poros razoavelmente grandes,
respectivamente. As isotermas do tipo III e V são características de sistemas onde as

7
moléculas do adsorvato apresentam maior interação entre si do que com o sólido, o que
representa um caso muito raro entre os materiais mais comuns.
Figura 3: Isotermas (volume adsorvido - n versus P/P0) do tipo I ao VI
Devido ao fenômeno de histerese, a isoterma do tipo IV e V apresentam dois valores
de pressão relativa para cada valor de quantidade adsorvida. A ocorrência disso é função do
formato do poro e do menisco do líquido. Para poro de formato cilíndrico fechado em uma
das extremidades a condensação capilar começa no fundo do poro, formando um menisco
hemisférico. A evaporação se inicia a partir desse menisco na mesma pressão que a
condensação ocorreu, gerando uma isoterma sem histerese.
2.3 Modelo de Langmuir
Langmuir apresentou uma teoria para entender a adsorção sobre uma superfície
uniforme, simples, infinita e não porosa.
O exemplo resume-se em uma hipótese de atividade das moléculas adsorvidas pela
superfície do adsorvente: assim que as moléculas são adsorvidas, existe uma divisão uniforme
gerando uma monocamada que recobre completamente a superfície. Neste modelo, utiliza-se
o conceito dinâmico de equilíbrio de adsorção que determina a igualdade das velocidades de
adsorção e dessorção.
Logo, assume-se os seguintes pressupostos:
● Existe um número definido de sítios.

8
● Os sítios têm energia equivalente e as moléculas adsorvidas não interagem
umas com as outras.
● A adsorção ocorre em uma monocamada.
● Cada sítio pode comportar apenas uma molécula adsorvida.
A equação a seguir representa a isoterma de Langmuir:
𝑞 = 𝑞𝑚𝑎𝑥 . 𝐾𝑙. 𝐶𝑒
1 + 𝐾𝑙 . 𝐶𝑒
Onde:
q: quantidade do soluto adsorvido por grama de adsorvente no equilíbrio (mg g-1);
qmax: capacidade máxima de adsorção (mg g-1);
Kl: constante de interação adsorvato/adsorvente (L mg-1);
Ce: concentração do adsorvato no equilíbrio (mg L-1).
Quando uma solução é posta em contato com o adsorvente e o sistema atinge o
equilíbrio, este estado de equilíbrio nada mais é do que a igualdade da velocidade em que as
moléculas ou íons são adsorvidos/dessorvidos na superfície do adsorvente. Assim, se a
velocidade de adsorção é proporcional à concentração do adsorvato no líquido (Ce), e para a
fração da área de superfície do adsorvente que está vazia (1- θ), onde θ é a fração da
superfície coberta, pode-se escrever a equação:
𝑇𝑎𝑥𝑎 𝑑𝑒 𝑎𝑑𝑠𝑜𝑟çã𝑜 = 𝑘1𝐶𝑒 (1 − 𝜃)
Onde:
k1: constante para adsorção.
Admitindo-se que todos os sítios da superfície do adsorvente possuem a mesma
energia, ou seja, são de tal forma homogêneos, k1 assume o mesmo valor para todos os sítios.
Além disso, admite-se que a cobertura da superfície se dá de maneira mono (molecular ou
elementar), isto é, somente é possível a formação de uma monocamada. Então, a taxa de
adsorção é proporcional a (1 - θ), isto é, a total cobertura (adsorção) estará completa quando θ
for igual a 1.
Da mesma forma como considerado com a taxa de adsorção, considerando que o
sistema se encontra em equilíbrio, tem-se para a taxa de dessorção, portanto:

9
𝑇𝑎𝑥𝑎 𝑑𝑒 𝑑𝑒𝑠𝑠𝑜𝑟çã𝑜 = 𝑘2𝜃
Onde:
k2: constante para a dessorção.
Como o sistema encontra-se em estado de equilíbrio, pode-se igualar as duas taxas:
𝑘1𝐶𝑒 (1 − 𝜃) = 𝑘2𝜃
ou, resolvendo para 𝜃 e adotando 𝐾𝐿 =𝑘1
𝑘2, tem-se que:
𝜃 =𝐾𝐿𝐶𝑒
1 + 𝐾𝐿𝐶𝑒
Em geral, é preferível trabalhar em termos da quantidade q, a quantidade de soluto
adsorvido por massa de adsorvente, em vez de θ. A equação da isoterma de Langmuir é
frequentemente rearranjada para outras formas lineares para determinar os valores de KL e
qmax, como mostrado nas equações abaixo:
1
𝑞𝑒=
1
𝑞𝑚𝑎𝑥+
1
𝐾𝐿𝑞𝑚𝑎𝑥𝐶𝑒
𝐶𝑒
𝑞𝑒=
1
𝑞𝑚𝑎𝑥𝐶𝑒 +
1
𝐾𝐿𝑞𝑚𝑎𝑥
𝑞𝑒 = 𝑞𝑚𝑎𝑥 − (1
𝐾𝐿)
𝑞𝑒
𝐶𝑒
𝑞𝑒
𝐶𝑒= 𝐾𝐿𝑞𝑚𝑎𝑥 − 𝐾𝐿𝑞𝑒
Geralmente, as duas primeiras formas são mais utilizadas. Portanto, tomando como
base a primeira delas, a construção do gráfico 1/q versus 1/Ce irá produzir uma linha reta (a
qual é geralmente obtida por um procedimento de ajuste linear por mínimos quadrados) com
inclinação 1/(KLqmax) e intersecção 1/qmax. Conhecendo-se esses valores, pode-se facilmente
calcular os dois parâmetros KL e qmax.
A medida de equilíbrio 𝑅𝑙 permite prever a configuração da isoterma de adsorção
(favorável, desfavorável) e pode ser calculada pela equação:
𝑅𝑙 = 1
1 + 𝐾𝑙𝐶0
Onde:
Co: concentração inicial na fase líquida (mg L-1).

10
Na maioria das situações de adsorção, o adsorvato prefere a fase sólida à líquida e a
adsorção é dita favorável, 0< RL< 1. Quando RL> 1, há o indicativo de que o soluto prefere a
fase líquida à sólida. RL=1 corresponde a uma isoterma linear e RL = 0 corresponde a uma
isoterma irreversível.
Aplicações das Isotermas de Langmuir
Conforme Linhares et al. (2008) escreveu, a capacidade dos coloides do solo em
adsorver metais pesados e mantê-los retidos com grande energia depende de muitos fatores.
Em geral é avaliada a capacidade de adsorção de cádmio e chumbo por diferentes solos e a
influência de seus constituintes, para prever a quantidade destes elementos.
Como já foi dito anteriormente, a quantidade de elemento adsorvido pela concentração
remanescente na solução de equilíbrio pode ser representada pelas isotermas de adsorção
(McBride, 1994). Comumente são utilizadas as isotermas de Langmuir e Freundlich para a
determinação da máxima retenção de metais pelos solos e suas energias associadas. Dentre
todos os modelos de isotermas, o que tornou a equação de Langmuir atrativa foi o
fornecimento de parâmetros quantitativo (máxima capacidade de adsorção) e qualitativo
(expressão da energia de ligação).
No trabalho de Linhares et al., 2008, os parâmetros das isotermas dos solos
investigados foram estimados por um programa de regressão não linear, a fim de minimizar os
erros provindos da linearização de dados.
De acordo com Harter (1984), no que se refere à equação de Langmuir, por exemplo,
o aspecto negativo proveniente do uso de sua forma linear, dá-se pelo fato de, ao plotar Ceq
contra Ceq/q, reduz-se a variabilidade dos dados, uma vez que Ceq é plotado contra si
mesmo. Além disso, nem sempre a adoção de formas lineares irá proporcionar curvas mais
bem ajustadas às isotermas obtidas experimentalmente. Dessa forma, efetuou-se o ajuste das
curvas de adsorção com base na forma original das equações de Langmuir e de Freundlich.
11
Além dos estudos de metais adsorvidos nos solos brasileiros, as isotermas de
Langmuir são usadas para a remoção de amônia dos efluentes de suinícolas.
Segundo Higarashi, Kunz e Mattei (2008), as isotermas de Langmuir podem descrever
as interações essenciais entre o adsorvente e o cátion a ser removido da solução (NH4+) como
forma de otimização para interpretar os dados de forma mais simples, para tratamento de
efluentes. Como a equação de isoterma de equilíbrio de Langmuir parte do pressuposto que
existe um número finito de sítios de ligação os quais se encontram homogeneamente
distribuídos sobre a superfície do adsorvente, possuindo igual afinidade de se ligar a uma
camada de moléculas as quais, por sua vez, não interagem entre si.
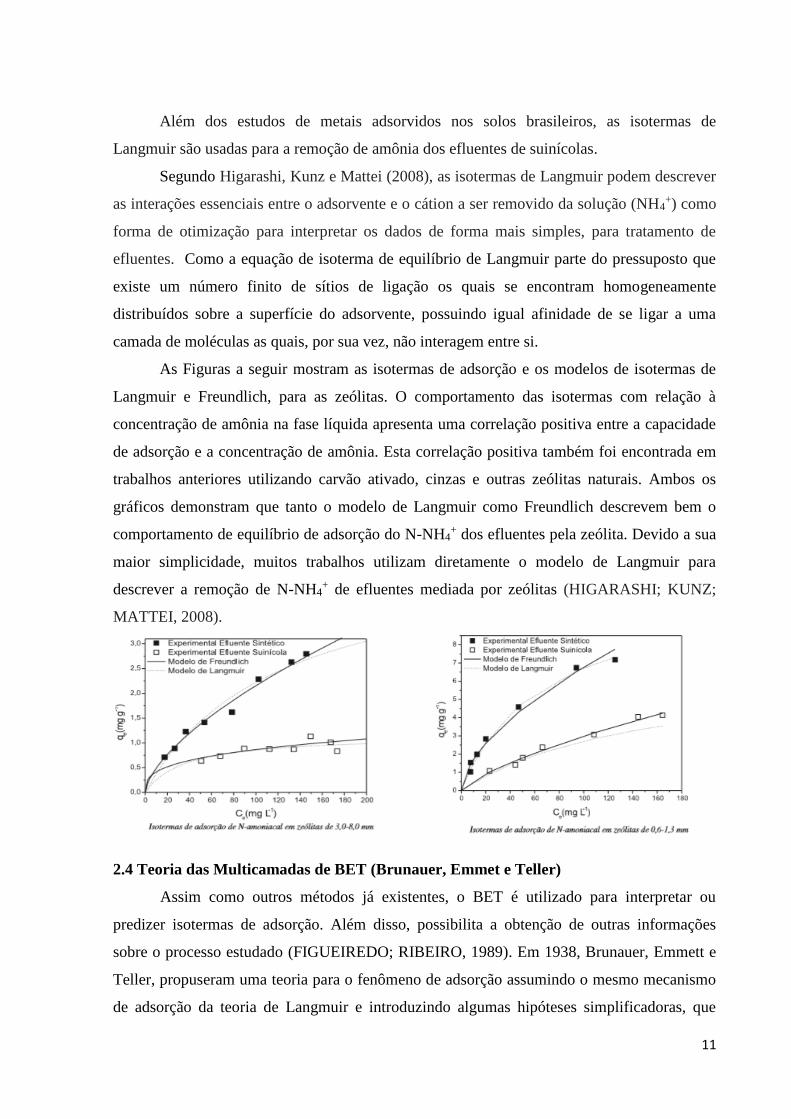
As Figuras a seguir mostram as isotermas de adsorção e os modelos de isotermas de
Langmuir e Freundlich, para as zeólitas. O comportamento das isotermas com relação à
concentração de amônia na fase líquida apresenta uma correlação positiva entre a capacidade
de adsorção e a concentração de amônia. Esta correlação positiva também foi encontrada em
trabalhos anteriores utilizando carvão ativado, cinzas e outras zeólitas naturais. Ambos os
gráficos demonstram que tanto o modelo de Langmuir como Freundlich descrevem bem o
comportamento de equilíbrio de adsorção do N-NH4+ dos efluentes pela zeólita. Devido a sua
maior simplicidade, muitos trabalhos utilizam diretamente o modelo de Langmuir para
descrever a remoção de N-NH4+ de efluentes mediada por zeólitas (HIGARASHI; KUNZ;
MATTEI, 2008).
2.4 Teoria das Multicamadas de BET (Brunauer, Emmet e Teller)
Assim como outros métodos já existentes, o BET é utilizado para interpretar ou
predizer isotermas de adsorção. Além disso, possibilita a obtenção de outras informações
sobre o processo estudado (FIGUEIREDO; RIBEIRO, 1989). Em 1938, Brunauer, Emmett e
Teller, propuseram uma teoria para o fenômeno de adsorção assumindo o mesmo mecanismo
de adsorção da teoria de Langmuir e introduzindo algumas hipóteses simplificadoras, que

12
admitem a possibilidade de que uma camada tenha a capacidade de produzir sítios de
adsorção gerando a deposição de uma camada sobre outra (SCHONS, 2010).
Figura 4: Representação figurativa do modelo de Langmuir e BET, respectivamente.
A teoria de Brunauer, Emmett e Teller é uma extensão dos fundamentos da teoria de
Langmuir para permitir uma adsorção maior, para a formação de duas ou mais camadas na
superfície. A teoria leva em consideração que as forças envolvidas na adsorção física são
similares àquelas envolvidas na liquefação, ou seja, força de Van der Waals, e que a adsorção
física não está limitada à formação de monocamada, mas pode continuar e formar
multicamadas sobre a superfície do adsorvente (NASCIMENTO et al., 2014). A equação,
denominada BET (letras iniciais dos nomes dos três autores), se baseia na hipótese de que as
forças responsáveis pela condensação do gás são também responsáveis pela atração de várias
moléculas para a formação de multicamadas. Brunauer, Emmett e Teller generalizaram a
equação de Langmuir considerando que a velocidade de condensação das moléculas da fase
gasosa sobre a primeira camada é igual à velocidade de evaporação da segunda camada
(TEIXEIRA; COUTINHO; GOMES, 2001).
O modelo BET pode ser representado pelas equações:
Em que:
Ct: constante relacionada com o tamanho do poro;
P: pressão;
Po: pressão de saturação do gás;
VA: quantidade de gás adsorvido (mL);
Vm: capacidade de adsorção na monocamada.

13
Em que:
C: concentração da espécie adsorvente;
q1: calor de adsorção da primeira camada;
qL: calor de liquefação do gás adsorvente.
Na forma linear, pode-se obter a equação abaixo.
Graficamente, o método pode ser demonstrado da seguinte forma:
Figura 5: Representação gráfica do método BET.
O modelo BET segue algumas considerações como a homogeneidade da superfície do
sólido, não contando com possíveis imperfeições geradas por impurezas e defeitos, causando
distorções no potencial da superfície. Não são consideradas também as forças horizontais
entre moléculas de uma mesma camada, somente interações verticais entre o adsorvente e a
molécula adsorvida. Além disso, todas as camadas, exceto a primeira, são tratadas de modo
equivalente, não tratando de um possível declínio do potencial atrativo gerado na superfície.
A entalpia de adsorção é igual à entalpia de condensação (SCHONS, 2010).
2.5 Isoterma de adsorção de Gibbs
A tensão superficial, 𝛾, é o trabalho necessário para aumentar a área de uma superfície,
por unidade de área. Ou seja,
𝛾 = 𝑑𝑊 . 𝑑𝐴

14
A unidade da tensão superficial, no SI, é joules por metro quadrado. Para
conseguirmos relacionar o trabalho com a concentração de uma espécie na superfície da
solução devemos imaginar a solução em duas partes: o interior e a superfície. Tanto no
interior quanto na superfície possuem potenciais químicos característicos. Portanto,
chamemos de 𝜇𝑖 o potencial químico do interior da solução e de 𝜇𝑠 o potencial químico da
superfície. O potencial químico é a Energia Livre de Gibbs relacionada à adição de uma
molécula em uma determinada fase.
Quando uma molécula do interior da solução migra para a superfície há um ganho de
energia livre. Esse ganho diminui o trabalho necessário para aumentar a área da superfície
desta molécula em uma quantidade equivalente. Desta forma nota-se uma relação entre a
tensão superficial do líquido na presença de um surfactante e a diferença de potencial químico
do surfactante na superfície relativamente ao seio da solução. Com pressão e temperatura
constantes, a variação da energia livre corresponde à variação dos potenciais químicos.
Portanto temos, quantitativamente,
𝛾𝑑𝐴 = 𝛾 𝑜 𝑑𝐴 − (𝜇𝑖 − 𝜇𝑠 )𝑑𝑛
Onde:
𝛾𝑜é a tensão superficial do líquido puro;
𝑑𝑛 é o número de moléculas que migraram do interior do líquido para a superfície 𝑑𝐴
a partir de uma distribuição homogênea;
𝛾𝑑𝐴 é o trabalho necessário para expandir a superfície em 𝑑𝐴;
(𝜇𝑠 − 𝜇𝑖)𝑑𝑛 é o trabalho realizado pelas 𝑑𝑛 moléculas que migraram do interior do
líquido para superfície.
Pela equação acima podemos perceber que o trabalho necessário para expandir a
superfície de líquido é igual ao trabalho que seria necessário no líquido puro menos a energia
livre que se ganha ao transferir moléculas do interior do líquido para a superfície.
A equação acima pode ser reescrita como:
(𝛾 − 𝛾𝑜 )𝑑𝐴 = −(𝜇𝑖 − 𝜇𝑠 )𝑑𝑛
O potencial químico é dado por
𝜇 = 𝜇𝑜 + 𝑅𝑇 𝑙𝑛 𝑎
Onde 𝜇 𝑜 é o potencial químico da substância puro e “a” é a atividade da substancia no
sistema em questão. Substituindo podemos chegar a:
(𝛾 − 𝛾 𝑜)𝑑𝐴 = −𝑅𝑇 𝑙𝑛 (𝑎𝑖
𝑎𝑠 ) 𝑑𝑛

15
Onde 𝑎𝑠 é a atividade do surfactante na superfície e ai é a atividade dele no interior da
solução. Rearranjando, temos:
𝛾 − 𝛾 𝑜 = −𝑅𝑇 𝑙𝑛 (𝑎𝑖
𝑎𝑠)
𝑑𝑛
𝑑𝐴 ,
Notamos que 𝑑𝑛/𝑑𝐴 é a quantidade de moléculas do surfactante que migrou do seio
da solução para a superfície a partir de uma distribuição homogênea, por unidade de área da
superfície. Denominaremos este valor como “concentração superficial excedente do soluto”
na superfície de Gibbs, que representa o excesso de moléculas próximo da superfície de Gibbs
para uma dada situação de equilíbrio, Г. Assim,
𝛾 − 𝛾 𝑜 = −𝑅𝑇 𝑙𝑛 (𝑎𝑖
𝑎𝑠) 𝛤
A superfície consegue acomodar uma quantidade limitada de moléculas de soluto.
Portanto, ela possivelmente se satura antes do seio da solução. Desta forma, em um quadro
em que a superfície se aproxima da saturação ela é tomada pelo soluto e a atividade dele na
superfície se aproxima da unidade; temos 𝑎𝑠 = 1.
Como se trata de uma solução diluída, a atividade pode ser aproximada em uma
concentração, 𝐶𝑖 e teremos:
𝛾 − 𝛾 𝑜 = −𝑅𝑇 𝑙𝑛(𝐶𝑖 )𝛤
Se a concentração do soluto no seio da solução varia de 𝐶1 para 𝐶2, a tensão
superficial varia de acordo:
𝛾2 − 𝛾1 = −𝑅𝑇 𝛤[ 𝑙𝑛(𝐶2 ) − 𝑙𝑛(𝐶1 )]
ou seja,
𝛥 𝛾 𝛥𝑙𝑛𝐶 = −𝑅𝑇 𝛤
para alterações pequenas de concentração, escrevemos
(𝜕 𝛾
𝜕𝑙𝑛 𝐶)
𝑇,𝑝 = −𝑅𝑇 𝛤
Devido às considerações no começo das contas, esta equação é válida quando a
pressão e temperatura não variam. Esta equação é conhecida como isoterma de Gibbs. A
isoterma dá, portanto, a variação da tensão superficial com a concentração do soluto em
função da adsorção, positiva ou negativa, do referido soluto na interface. Se o soluto tiver
uma adsorção de superfície positiva em relação ao solvente (sua proporção em relação ao
solvente é maior do que dentro da fase), a tensão superficial diminui à medida que a
concentração aumenta. Se, por outro lado, o soluto tiver uma adsorção superficial negativa,
Γ<0 a tensão superficial aumentará com a concentração.

16
Em geral, se o solvente (componente 1) é água, três tipos de comportamento podem
ser observados para a tensão superficial como uma função da concentração de soluto
(componente 2), dependendo da natureza deste último, como mostrado na figura.
Figura 6: Variação da tensão superficial de soluções aquosas em função da
concentração de diferentes substâncias.
1) Tipo I ou substâncias inativas. Para estas substâncias, a tensão superficial
aumenta ligeiramente com sua a concentração (𝜕 𝛾
𝜕𝑙𝑛 𝐶) >0 e assim de acordo
com a equação da isoterma de Gibbs, 𝛤2< 0. Ou seja, são substâncias que não
têm tendência a adsorver na interface, mas, pelo contrário, interagem
fortemente com as moléculas do solvente (razão pela qual se acumulam no
interior da fase aquosa). Devido às interações com moléculas de solvente (por
exemplo, íon-dipolo), sua situação de energia será mais estável na dissolução
do que na superfície, uma vez que, um aumento na área de superfície exigirá
mais trabalho para trazer moléculas do interior para a interface. A tensão
superficial aumenta em relação ao solvente puro, e quanto maior a
concentração do soluto, como mostrado na figura anterior. No caso da água, as
substâncias inativas são sais inorgânicos (NaCl).
2) Para as substâncias dos tipos II e III, a tensão superficial diminui com a sua
concentração (𝜕 𝛾
𝜕𝑙𝑛 𝐶) <0. Estás são substâncias que tendem a adsorver na
interface, mostrando uma concentração maior em relação ao solvente na
interface do que dentro da fase aquosa. De particular interesse estão as

17
substâncias que possuem uma parte polar ou hidrofílica (por exemplo, grupos -
OH, -COOH) e uma parte não polar ou hidrofóbica (cadeias de
hidrocarbonetos), pois enquanto a parte polar interage fortemente com
moléculas de água, aumentando a solubilidade, a parte de hidrocarbonetos irá
interagir fracamente com as moléculas de água. A parte de hidrocarboneto
tenderá a estar localizada fora do solvente, pois desta forma não quebra as
interações H2O-H2O que são fortes (pontes de hidrogênio). Assim, Γ2> 0. A
tensão superficial diminui desde então, pois para aumentar a superfície as
moléculas essencialmente de soluto seriam colocadas na interface e como estas
moléculas interagem fracamente com o solvente seria menos difícil levá-las do
interior da fase aquosa para a interface, tornando a tensão superficial menor.
Dessa forma o comportamento II ou III é determinado pela insolubilidade da
substância, por exemplo, o comprimento da cadeia de hidrocarbonetos. Quando é pequeno
(etanol, ácido acético), o composto é parcialmente solúvel em água e acumula-se lentamente
na interface, dando origem a uma evolução do tipo II.
Se a cadeia de hidrocarbonetos é grande (com 10 ou mais átomos de carbono), estas
moléculas são muito pouco solúveis e se acumulam rapidamente na superfície, os agentes
tensoativos também chamados de surfactantes são exemplos desses compostos que dão uma
evolução do tipo III (Γ aumenta rapidamente com a concentração, de modo que, de acordo
com a isoterma de Gibbs, γ rapidamente diminui com ela). A tensão superficial deve variar
linearmente com o logaritmo da concentração do soluto do tipo III na solução, nas
proximidades da saturação da superfície. Quando a saturação é alcançada, a tensão superficial
não deve mais variar, permanecendo relativamente constante. O gráfico de tensão superficial
em função do logaritmo da concentração deve assemelhar, nestas condições, ao gráfico
abaixo.
Para concentrações abaixo da concentração de saturação da superfície, a tensão
superficial deve convergir para a tensão superficial do solvente puro.

18
Figura 7 - Variação da tensão superficial de um líquido em função da
concentração de surfactante (ou qualquer soluto cujo potencial químico na superfície é
menor que no seio da solução).
Muitos parâmetros relevantes podem ser conseguidos a partir desta curva.
Primeiramente, faz-se clara a concentração de surfactante a partir da qual a superfície está
saturada. Se considerarmos que uma vez que as moléculas de surfactante não podem
preencher a superfície elas se aglomeram no seio da solução, formando micelas, esta é a
concentração desde a qual a formação de micelas é um processo propício. A esta concentração
intitulamos de Concentração Micelar Crítica (CMC). Aqui não demonstramos que se formam
micelas no seio da solução, a prova deste fenômeno requer experimentos independentes.
Nas proximidades da CMC, a tensão superficial reduz com a concentração na taxa
−𝑅𝑇Г. Г é o número de moléculas em excesso na superfície, em relação ao seio da solução,
por unidade de área. Se o surfactante é pouco solúvel (e suas moléculas se concentram na
superfície muito preferencialmente), praticamente não há moléculas de surfactante no seio da
solução até a CMC, portanto Г é simplesmente a densidade superficial de surfactante (o
número de surfactantes por unidade de área).
3. APLICAÇÕES
3.1 Adsorção de cádmio e chumbo em diferentes classes de solos brasileiros – Aplicação
dos modelos de Langmuir e Freundlich
Conforme Linhares et al. (2008) escreveu, a capacidade dos coloides do solo em
adsorver metais pesados e mantê-los retidos com grande energia depende de muitos fatores.

19
Em geral é avaliada a capacidade de adsorção de cádmio e chumbo por diferentes solos e a
influência de seus constituintes, para prever a quantidade destes elementos.
Como já foi dito anteriormente, a quantidade de elemento adsorvido pela concentração
remanescente na solução de equilíbrio pode ser representada pelas isotermas de adsorção
(McBride, 1994). Comumente são utilizadas as isotermas de Langmuir e Freundlich para a
determinação da máxima retenção de metais pelos solos e suas energias associadas. Dentre
todos os modelos de isotermas, o que tornou a equação de Langmuir atrativa foi o
fornecimento de parâmetros quantitativo (máxima capacidade de adsorção) e qualitativo
(expressão da energia de ligação).
No trabalho de Linhares et al., 2008, os parâmetros das isotermas dos solos
investigados foram estimados por um programa de regressão não linear, a fim de minimizar os
erros provindos da linearização de dados.
De acordo com Harter (1984), no que se refere à equação de Langmuir, por exemplo,
o aspecto negativo proveniente do uso de sua forma linear, dá-se pelo fato de, ao plotar Ceq
contra Ceq/q, reduz-se a variabilidade dos dados, uma vez que Ceq é plotado contra si
mesmo. Além disso, nem sempre a adoção de formas lineares irá proporcionar curvas mais
bem ajustadas às isotermas obtidas experimentalmente. Dessa forma, efetuou-se o ajuste das
curvas de adsorção com base na forma original das equações de Langmuir e de Freundlich.
Além dos estudos de metais adsorvidos nos solos brasileiros, as isotermas de
Langmuir são usadas para a remoção de amônia dos efluentes de suinícolas.
Segundo Higarashi, Kunz e Mattei (2008), as isotermas de Langmuir podem descrever
as interações essenciais entre o adsorvente e o cátion a ser removido da solução (NH4+) como
forma de otimização para interpretar os dados de forma mais simples, para tratamento de
efluentes. Como a equação de isoterma de equilíbrio de Langmuir parte do pressuposto que
existe um número finito de sítios de ligação os quais se encontram homogeneamente

20
distribuídos sobre a superfície do adsorvente, possuindo igual afinidade de se ligar a uma
camada de moléculas as quais, por sua vez, não interagem entre si.
As Figuras a seguir mostram as isotermas de adsorção e os modelos de isotermas de
Langmuir e Freundlich, para as zeólitas. O comportamento das isotermas com relação à
concentração de amônia na fase líquida apresenta uma correlação positiva entre a capacidade
de adsorção e a concentração de amônia. Esta correlação positiva também foi encontrada em
trabalhos anteriores utilizando carvão ativado, cinzas e outras zeólitas naturais. Ambos os
gráficos demonstram que tanto o modelo de Langmuir como Freundlich descreve bem o
comportamento de equilíbrio de adsorção do N-NH4+ dos efluentes pela zeólita. Devido a sua
maior simplicidade, muitos trabalhos utilizam diretamente o modelo de Langmuir para
descrever a remoção de N-NH4+ de efluentes mediada por zeólitas (HIGARASHI; KUNZ;
MATTEI, 2008).
3.2. Área Específica de catalisadores
Um dos métodos mais comuns de determinação da área específica de um sólido se baseia
na determinação da quantidade de um adsorvato necessária para recobrir com uma
monocamada a superfície de um adsorvente. Os adsorbatos normalmente utilizados para esse
fim são gases e, por isso, torna-se necessário o estudo da interação entre o gás e o sólido no
processo de adsorção.
Além disso, destaca-se aqui importância de se determinar a distribuição de tamanhos de
poros para um sólido e nesse caso, catalisadores, o qual é um parâmetro muito importante
para o estudo da estrutura porosa, já que está intimamente relacionado à área total do sólido.
Assim, devemos observar que pelos pontos apresentados, caracterizar as propriedades
texturais dos catalisadores heterogêneos é com certeza umas das principais funções do

21
profissional de catálise, a qual usa diariamente conceitos de adsorção física e química. Isso é
fato, porque a relação entre área do catalisador disponível como sítio ativo é o principal fator
que contribui para a vida útil dele, por isso a importância de se determinar esses parâmetros.
Adsorção de nitrogênio
A distribuição de tamanhos ou de volumes de poro em função do diâmetro de poro
pode ser calculada a partir da pressão relativa na qual os poros são preenchidos com um
líquido proveniente da condensação de um gás. O processo inverso, ou seja, a evaporação do
líquido contido no poro, também pode ser utilizado.
Um exemplo de aplicação disso é na área de desenvolvimento aeroespacial, onde
alumina porosa é largamente utilizada como suporte para catalisadores heterogêneos para
suportar metais nobres como Irídio e Rutênio
As áreas superficiais específicas do suporte e do catalisador pronto (suporte mais
metal ativo) são determinadas, além de que se faz possível estudar os precursores do suporte,
Gibsita e Boehmita, por exemplo, para se ter controle no processo de obtenção da alumina
final como suporte. Para tais análises, utiliza-se um aparelho, como o da Figura 4.
Antecedendo a cada medida de adsorção, uma limpeza na superfície do material sempre deve
ser feita, processo que leva o nome de ativação de superfície - se aquece o analito a 473 K por
2 h, sob vácuo primário (10-3 bar).
Os valores das áreas superficiais específicas são assim obtidos através da adsorção de
N2, realizada à temperatura do nitrogênio líquido (77K e a 1 atm), segundo o método
desenvolvido por Brunauer et al. (1938).
Figura 4 – Aparelho Quantachrome utilizado para se rodar análises a base de adsorção
de gás Nitrogênio - INPE Cachoeira Paulista

22
REFERÊNCIAS
ATIKINS P; PAULA J; Físico-Química, 9ª, Rio de Janeiro ed. LTC, 2013.
FIGUEIREDO, J. L.; RIBEIRO, F. Ramôa. Catálise Heterogênea. Lisboa: Fundação
Calouste Gulbenkian, 1989.
MARTINEZ L. Introdução à Tensão Superficial, Instituto de Química Unicamp, 2012.
Disponível em:
http://leandro.iqm.unicamp.br/leandro/shtml/didatico/apoio/tensao_superficial_apoio.pdf
Müller, Carla Cristine; Rodriguez, Maria Teresa Raya-Rodriguez; Cybis, Luiz Fernando.
Adsorção em carvão ativado em pó para remoção de microcistina de água de
abastecimento público. Apostila PUC-Rio. Certificação Digital 0511121/CA.
LINHARES, Lucília Alves et al. Aplicação dos modelos de Langmuir e Freundlich na
adsorção de cádmio e chumbo em diferentes classes de solos brasileiros. Revista
Tecnológica, Belo Horizonte, Mg - Brasil, v. 17, n. 01, p.49-60, jan. 2008.
HIGARASHI, Martha Mayumi; KUNZ, Airton; MATTEI, Rosemari Martini. APLICAÇÃO
DE ADSORÇÃO PARA REMOVER AMÔNIA DE EFLUENTES SUINÍCOLAS PRÉ-
TRATADOS. Quim. Nova, Concórdia-SC, Brasil, v. 31, n. 5, p.1156-1160, jul. 2008.
NASCIMENTO, Ronaldo Ferreira et al. Adsorção: aspectos teóricos e ambientais.
Fortaleza: Imprensa Universitária, 2014.
SCHONS, Elenice. Fenômenos Interfaciais. Catalão: Elenice Schons, 2010. 39 slides, color.
TEIXEIRA, Viviane Gomes; COUTINHO, Fernanda M. B.; GOMES, Ailton S. Principais
Métodos De Caracterização Da Porosidade De Resinas À Base De Divinilbenzeno. Química
Nova, São Paulo, v. 24, n. 6, p.1-2, nov. 2001. Mensal. Disponível em:
<http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0100-40422001000600019>.
Acesso em: 01 abr. 2001.


















