
I Módulo 801 Problema 09 I Thiago Almeida Hurtado 1 ...
27
I Módulo 801 – Problema 09 I Thiago Almeida Hurtado 1) Revisar a anatomia da hipófise e hipotálamo, a fisiologia dos hormônios da hipófise anterior e sua interação com o hipotálamo e glândulas endócrinas 1) Introdução • O hipotálamo é composto por diversos neurônios, os quais regulam a função hipofisária, podendo levar a sintomas de hipopituitarismo. o Os núcleos médio-basais contêm os neurônios secretores de fatores hipofisiotróficos e os neurônios do centro da saciedade. Estes núcleos são próximos dos núcleos supraóptico e paraventricular que produzem ocitocina e ADH. ▪ Portanto, uma lesão hipotalâmica pode comprometer todos esses núcleos, levando ao pan- hipopituitarismo, ao diabetes insipidus central e, eventualmente, a obesidade hipotalâmica (por hiperfagia patológica). • A hipófise é uma glândula pequena (mede aproximadamente 1cm 2 e pesa 0,5 gramas), localizada na sela túrcica, que secreta diversos hormônios os quais estimulam as glândulas endócrinas periféricas (tireoide, suprarrenal, gônadas...). o É dividida anatomicamente em adeno-hipófise (anterior) e neuro-hipófise (posterior). o Suas relações anatômicas são importantes para a compreensão de sinais e sintomas decorrentes de lesões expansivas dessa estrutura. ▪ Superiormente: diafragma da sela (deflexão da dura-máter), protege contra a pressão do espaço subaracnoide. ▪ Anteriormente: quiasma óptico (possível acometimento da visão) ▪ Lateralmente: seios cavernosos, sifão carotídeo e pares cranianos (III, IV, VI e V) • De modo geral, a regulação hormonal da hipófise anterior apresenta três níveis de controle o Primeiro nível: hormônios atingem a hipófise, via sistema porta, exercendo feedback sob o próprio órgão
Transcript of I Módulo 801 Problema 09 I Thiago Almeida Hurtado 1 ...

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
1) Revisar a anatomia da hipófise e hipotálamo, a fisiologia dos hormônios da hipófise anterior e sua
interação com o hipotálamo e glândulas endócrinas
1) Introdução
• O hipotálamo é composto por diversos neurônios, os quais regulam a função hipofisária, podendo levar a
sintomas de hipopituitarismo.
o Os núcleos médio-basais contêm os neurônios secretores de fatores hipofisiotróficos e os
neurônios do centro da saciedade. Estes núcleos são próximos dos núcleos supraóptico e
paraventricular que produzem ocitocina e ADH.
▪ Portanto, uma lesão hipotalâmica pode comprometer todos esses núcleos, levando ao pan-
hipopituitarismo, ao diabetes insipidus central e, eventualmente, a obesidade hipotalâmica
(por hiperfagia patológica).
• A hipófise é uma glândula pequena (mede aproximadamente 1cm2 e pesa 0,5 gramas), localizada na sela
túrcica, que secreta diversos hormônios os quais estimulam as glândulas endócrinas periféricas (tireoide,
suprarrenal, gônadas...).
o É dividida anatomicamente em adeno-hipófise (anterior) e neuro-hipófise (posterior).
o Suas relações anatômicas são importantes para a compreensão de sinais e sintomas decorrentes
de lesões expansivas dessa estrutura.
▪ Superiormente: diafragma da sela (deflexão da dura-máter), protege contra a pressão do
espaço subaracnoide.
▪ Anteriormente: quiasma óptico (possível acometimento da visão)
▪ Lateralmente: seios cavernosos, sifão carotídeo e pares cranianos (III, IV, VI e V)
• De modo geral, a regulação hormonal da hipófise anterior apresenta três níveis de controle
o Primeiro nível: hormônios atingem a hipófise, via sistema porta, exercendo feedback sob o
próprio órgão

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
o Segundo nível: secreção de de citocinas e fatores de
crescimento no interior da própria hipófise,
regulando a sua função por sinalização parácrina e
autócrina.
o Terceiro nível: exercido pelos hormônios periféricos
sob a hipófise (em geral, feedback negativo sob a
síntese de hormônios que estimulam sua produção)
• As doenças da hipófise e do hipotálamo podem levar a
estados de hipo ou hiperfunção hormonal.
2) Adeno-Hipófise (Pituitária Anterior)
• Essa estrutura contém células especializadas na síntese e liberação de hormônios peptídicos.
• A função das células da adeno-hipófise é controlada por neurônios hipotalâmicos especializados,
localizados na porção médio-basal do hipotálamo.
• O pedículo hipofisário é de extrema importância para o bom funcionamento dessa estrutura, visto
que ele abriga o sistema porta, através do qual os hormônios hipotalâmicos atingem a hipófise.
o ATENÇÃO! O comprometimento do pedículo leva a queda de todos os fatores liberadores (GH,
TSH, ACTH, LH e FSH). A única excessão é a prolactina, a qual perde o seu estímulo inibitório
(DOPAMINA) e sua secreção é estimulada!
• Abaixo, uma tabela resumindo os hormônios, o tipo celular na hipófise que os secreta e sua função:
Hormônio Tipo Celular Função
GH Somatotrofo - Produção de IGF
- Regula metabolismo de
carboidratos, proteínas e
lipídeos
- Promove anabolismo e
crescimento
Prolactina Lactotrofo - Desenvolvimento mamário
- Lactação
TSH Tireotrofo - Estimula secreção de T3 e
T4 por parte da tireoide
ACTH Corticotrofo - Estimula secreção de
cortisol
- Estimula secreção de
andrógenos
- Estimula secreção de
aldosterona (em menor nível)
LH Gonadotrofo - Estimula produção de
testosterona
- Estimula ovulação
FSH Gonadotrofo - Estimula crescimento
testicular
- Estimula espermatogênese
- Estimula geração de
folículos ovarianos

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
a) Hormônio do crescimento/somatotrofinas (GH)
• Células: somatotrofos, as mais numerosas da hipófise (~50% da massa celular)
• Estímulo hipotalâmico: GHRH, hormônio produzido pelo núcleo arqueado do hipotálamo
o O estrógeno causa aumento dos receptores desse hormônio nos somatotrofos, otimizando a sua
ação e a consequente síntese e secreção de GH.
• O GH apresenta meia vida de 20 minutos e sua secreção é pulsátil (sendo sua secreção maior e mais
frequente à noite/madrugada). Esses pulsos tendem a aumentar durante a puberdade e reduzir após a
terceira década de vida e com o envelhecimento.
o Aumento da síntese de GH: jejum, hipoglicemia, grelina, exercício físico, diabetes,
hipertireoidismo.
o Redução da síntese de GH: GH e IGF-1 (feedback negativo), hipercortisolismo crônico,
hipotireoidismo, hiperglicemia pós-prandial, obesidade
• O GH exerce seu efeito principalmente ao se ligar a receptores hepáticos, estimulando a produção de
IGF (fatores de crescimento semelhantes à insulina), os quais irão mediar os efeitos sistêmicos do GH.
Os estrógenos orais reduzem a produção de IGF, enquanto os andrógenos a estimulam.
o Aumento da síntese de IGF: puberdade, gravidez, hipertireoidismo e acromegalia
o Redução da síntese de IGF: desnutrição, cirrose ou hepatite ativa, hipotireoidismo, insuficiência
cardíaca, AIDS, reposição VO de estrógeno
• O GH e o IGF-1 agem promovendo o crescimento e regulando o metabolismo de carboidratos, proteínas,
lipídeos e o metabolismo mineral ósseo.
• Em excesso, o GH provoca resistência insulínica, levando ao aumento da produção hepática de glicose e
à menor oxidação e captação da glicose por tecidos periféricos.
b) Somastotatina
• A somatostatina é uma proteína produzida em várias partes do corpo, inibindo a proliferação e o
crescimento das células somatotróficas.
c) Prolactina
• Células: lactotrofos (10 a 20% das células, podendo chegar a 50% na gestação – hiperplasia da hipófise
durante a gestação e amamentação)
• “Estímulo” hipotalâmico: É estimulado em parte pelo TRH (sem fator específico). A dopamina é seu
principal inibidor.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Sua função é basicamente permitir o desenvolvimento mamário, a lactação nos mamíferos e estimular o
comportamento do instinto materno.
• A regulação de sua síntese e secreção é feita principalmente
pela via de inibição dopaminérgica. A dopamina produzida
no hipotálamo inibe a secreção e produção de prolactina, além
de inibir a proliferação dos lactotrofos.
• ATENÇÃO! Concentrações excessivas de prolactina
bloqueiam a síntese e a liberação de GnRH hipotalâmico, o que
leva a amenorreia e alteração de espermatozoides em pacientes
com hiperprolactinemia.
• ATENÇÃO! O estrógeno, ao contrário do que o corre no GH,
estimula a secreção e síntese da prolactina!
d) Hormônio adrenocorticotrófico (ACTH)
• Células: corticotrofos (cerca de 20% das células da adeno hipófise – produzem a POMC).
• Estímulo hipotalâmico: CRH, produzido no núcleo
paraventricular.
• O ACTH se liga principalmente ao córtex da adrenal,
onde estimula as camadas fasciculada e reticulada a
secretarem cortisol e andrógenos principalmente.
Também exerce um pequeno estímulo sob a secreção
de aldosterona.
• O ACTH e o cortisol são secretados conforme o ritmo
circadiano, com pico pela manhã e declínio ao longo do
dia.
• ATENÇÃO! O principal hormônio inibidor da síntese
e liberação tanto do CRH hipotalâmico quanto do
ACTH hipofisário é o próprio cortisol, através de
feedback negativo.
e) Gonadrotrofinas (LH e FSH)
• Células: gonadotrofos (correspondem a aproximadamente 10% das células)
• Estímulo hipotalâmico: GnRH, produzido pelo núcleo arqueado do hipotálamo
o Estimulam a síntese de GnRH: catecolaminas, kisspeptina, serotonina, MSH, TNF-alfa
o Reduzem a síntese de GnRH: PRL, opioides, GABA, betaendorfinas

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• FSH: estimula o crescimento testicular e espermatogênese, nas
mulheres estimula a geração de folículos ovarianos e a
aromatização de andrógenos.
• LH: estimula a produção de testosterona e em mulheres induz a
ovulação.
• ATENÇÃO! A secreção das gonadotrofinas depende da
secreção pulsátil do GnRH (caso secretado constantemente e
não em pulsos, as gonadotrofinas não são produzidas e nem
secretadas). Pulsos de GnRH a cada 1 hora estimulam a
produção de LH e pulsos a cada 3/4 h favorecem a produção de
FSH.
o A pulsatilidade ocorre até os 12 meses de idade
(minipuberdade) durante o primeiro ano de vida,
depois, só volta a ocorrer na puberdade, onde há reativação do eixo gonadotrófico.
• ATENÇÃO! O LH e o FSH têm sua produção inibida pelo hormônio inibina, que é produzido pelas
gônadas. É um hormônio produzido principalmente pelos folículos ovarianos e pelas células de Sertoli,
por isso sua dosagem é um marcador da integridade desses órgãos.
• ATENÇÃO! Os esteroides sexuais apresentam feedback negativo sob a produção de GnRH (exceto o
estradiol, quando sua exposição é prolongada e está em altas concentrações – importante para o pico de
LH no meio do ciclo ovulatório)
f) Tireotropina (TSH)
• Células: tireotrofos (5% das células da adeno-hipófise)
• Estímulo hipotalâmico: TRH, produzido pelos núcleos medial
e paraventricular do hipotálamo.
• É um hormônio proteico, composto por duas subunidades. A
subunidade alfa é a mesma que compõe as moléculas de LH,
FSH e HCG (a beta é específica para o TSH). Também
apresenta secreção pulsátil.
• O TSH se liga nas células foliculares da tireoide, onde
estimulam a glândula a produzir e secretar T3 e T4.
Perifericamente a T4 é convertida em T3.
• O TSH é inibido por T3, somastotatina, GH, dopamina,
desnutrição, anorexia, infecção e inflamação.
• ATENÇÃO! A insuficiência adrenal causa elevação aguda de
TSH.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
3) Hipófise posterior (neuro-hipófise)
• Consiste em um tecido neural, composto apenas de axônios distais de neurônios originados no
hipotálamo. Portanto, não contém células!
o Esses axônios contêm pacotes de hormônios armazenados para posterior liberação.
• A neuro-hipófise, ao contrário da hipófise anterior, apresenta irrigação diretamente das artérias
hipofisárias posterior (a adenohipófise é irrigada pelo sistema porta hipofisário).
• Secreta só dois hormônios: ADH e Ocitocina
a) Vasopressina (ADH)
• Na realidade, é produzido no hipotálamo na forma de pró-ADH, sendo transportado pela haste
hipofisária até a hipófise posterior, onde será clivado em ADH e neurofisinas 2 (responsáveis pelo
brilho da hipófise na RM e carreiamento de ADH).
• É responsável pela regulação da concentração da quantidade plasmática de sódio (natremia).
• No organismo, liga-se a três receptores, presentes na hipófise anterior, rins e vasos sanguíneos:
o Receptor V1: presente no músculo liso vascular, estimula a vasoconstrição e a agregação
plaquetária.
o Receptor V2: presentes nos túbulos coletores renais, estimulando a reabsorção de água
(expressão das aquoporinas e transporte por mecanismo de contracorrente)
o Receptor V3: presente na hipófise anterior nos corticotrofos, estimulando a secreção de ADH
• O principal fator que regula a secreção de ADH é a osmolaridade sérica, a qual é percebida
através de osmorreceptores. Quando a osmolaridade passa de 280 mOsm/kg, a secreção desse
hormônio é estimulada até um “nível máximo” (isso ocorre por volta de 295 mOsm/kg), no qual o
mecanismo da sede passa a ser o fator mais importante para o controle da osmolaridade.
o OBS: a PA e a volemia sanguínea também apresentam efeito sob a secreção de ADH, contudo,
tem um papel menor do que a osmolaridade sanguínea (barorreceptores no arco aórtico e no seio
carotídeo).
b) Ocitocina
• Seus principais efeitos são obstétricos: permite a contração do miométrio e contração da musculatura
lisa da mama (importante para ejeção do leite na amamentação).
• Também apresentam um efeito no sistema nervoso central, relacionado ao comportamento maternal.
• O principal estímulo para sua liberação é a sucção mamária. O estrógeno também estimula sua
liberação
2) Compreender o quadro clínico apresentado pelo paciente: pan-hipopituitarismo
1) Introdução

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• O termo hipopituitarismo se refere à redução de um ou mais hormônios hipofisários decorrente de
condições que acometem a região hipotálamo-hipofisária (o termo pan-hipopituitarismo é reservado para
condições em que há redução de todos os hormônios hipofisários ao mesmo tempo)
• É uma condição rara, com incidência de 4,2 em 100 mil pessoas/ano e prevalência de 45 em 100 mil
pessoas.
• Apresenta diversas etiologias, e quadro clínico variável (depende do tipo celular mais acometido e da
consequente deficiência hormonal). Costuma ser um quadro subdiagnosticado e de diagnóstico tardio
devido a inespecificidade das manifestações clínicas.
o Na criança, costuma se manifestar por déficit de crescimento
o No adulto, costuma se manifestar pelo hipogonadismo
o Graus variáveis de hipotireoidismo e hipoadrenalismo podem aparecer, podendo levar a
manifestações clínicas exuberantes devido a sua gravidade.
2) Etiologia
• É um evento causado pela destruição da adeno-hipófise ou pela deficiência de estímulos hipotalâmicos
que normalmente atuam sobre a hipófise (como por exemplo, lesões no hipotálamo médio basal ou no
pedículo hipfisário).
• Podemos dividir as etiologias em: doenças hipotalâmicas e doenças hipofisárias (hipopituitarismo
hipotalâmico ou hipofisário)
Hipopituitarismo Hipotalâmico
- Embriopatias congênitas da linha mediana: acometimento de fibras que ligam os dois hemisférios
cerebrais e dos tratos ópticos e olfativo. Assosiam-se ao lábio leporino e à fenda palatina.
- Síndrome de Prader-Willi: distúrbio relacionado a microdeleções no cromossomo 15. Manifesta-se
por hipotonia muscular nos primeiros meses de vida, evoluindo com obesidade hipotalâmica
(hiperfagia), baixa estatura, hipogonadismo hipogonadotrófico e alterações craniofaciais. Predominam
as deficiências de GnRH e GH.
- Síndrome de Bardet-Biedl: doença autossômica recessiva rara que se manifesta por obesidade
hipotalâmica, retinite pigmentosa, polidactilia e malformações renais.
- Síndrome de Kallman: distúrbio genético ligado ao X, caracterizado por anosmia/hiposmia (falha da
migração neuronal para o bulbo olfatório e hipotálamo), associada ao hipogonadismo
hipogonadotrófico. Manifesta-se por alta estatura e envergadura, micropênis, voz fina e testículos pré-
puberais.
- Adenomas hipofisários: acometem o hipotálamo por expansão
- Craniofaringioma: tumor comum em crianças, explicado abaixo (tumores selares e parasselares)
- Sarcoidose: invasão hipotalâmica dos grânulos sarcoides
- Outros: radioterapia, trauma cranioencefálcio, síndrome X

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
Hipopituitarismo Hipofisário
- Adenoma Hipofisário: o tumor pode comprimir a glândula funcionante, levando a redução
progressiva de sua secreção hormonal. Além disso, como já citado, podem se estender para o
hipotálamo, levando ao hipopituitarismo hipotalâmico.
- Síndrome da Sela Vazia: entidade comum no sexo feminino, marcada por um defeito no diafragma
da sela túrcica, que permite a herniação da membrana aracnoide para a fossa hipofisária. Isso leva ao
achatamento da hipófise no assoalho da célula e estiramento do pedículo hipofisário (o que explica a
hiperprolactinemia). Pode ser secundária a cirurgia, radioterapia ou infarto hipofisário.
- Apoplexia hipofisária: definida como uma hemorragia da hipófise que evolui para necrose, levando a
um quadro de pan-hipopituitarismo súbito. Geralmente ocorre devido a sangramento de tumor
hipofisário (secundário a hipertensão, diabetes). O quadro clínico é marcado por uma forte cefaleia,
associada a náuseas e vômitos. O paciente evolui rapidamente com queda do estado de consciência,
choque refratário à reposição volêmica e hiponatremia grave.
- Síndrome de Sheehan: causa de pan-hipopituitarismo no período puerperal.
a) Causas genéticas
• Geralmente se manifestam na infância, podendo causar deficiência isolada ou combinada de hormônios
hipofisários. São inúmeras as mutações que podem dar origem ao quadro de hipopituitarismo.
o ATENÇÃO! Algumas mutações genéticas, relacionadas a fatores de transcrição expressos no
período embrionário, podem levar a quadros de pan-hipopituitarismo por má-formação da
hipófise! Pode, inclusive, levar a málformações em outras áreas do SNC, gerando alterações
concomitantes ao pan-hipopituitarismo, como: displasia septo-óptica, neuro-hipófise ectópica,
rigidez de coluna cervical, malformações de Chiari (defeito congênito que acomete a conexão
cervical, levando a uma protusão posterior, a mais comum é a do tipo 2, geralmente vista em
crianças com espinha bífida, em que há o deslocamento da parte posterior do cérebro para baixo
através da parte inferior do crânio – pode haver hidrocefalia devido a obstrução do fluxo de
fluido cerebroespinhal), agenesia renal (síndrome de Kallman).
• O hormônio mais acometido nessa etiologia é o GH, o que leva a baixa estatura do indivíduo (déficit de
crescimento).
b) Tumores selares e parasselares
• Os principais são os adenomas hipofisários, neoplasias bem diferenciadas originadas da
adenohipófises. Podem ser divididos em funcionantes (3/4 dos casos – secretam hormônios) e não
funcionantes (1/4 dos casos). O tipo mais comum é o prolactinoma.
• Os microadenomas (tumor < 10 mm) não costumam causar insuficiência hipofisária diretamente.
• Os macroadenomas (tumor > 10 mm) hipofisários e outros tipos de tumores selares podem causar
hipopituitarismo pelo efeito compressivo sob o tecido hipofisário ao redor do tumor.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• ATENÇÃO! O craniofaringioma é o tumor mais comum dessa região em crianças. É o terceiro tipo de
tumor mais frequente do SNC nessa faixa etária. Manifesta-se com sinais de lesão expansiva do SNC
(cefaleia, vômitos, distúrbios visuais), poliúria (diabetes insipidus – 20% dos casos) e pan-
hipopituitarismo (50-75% dos casos). Nas crianças, manifesta-se por déficit de crescimento (em adultos
o quadro é de hipogonadismo). Sua característica mais marcante é a sua enorme tendência à
calcificação, a qual é visível na TC ou RNM de região selar. O tratamento é feito através da cirurgia
de ressecção, associada ou não a radioterapia e o prognóstico são bons.
Adenomas Hipofisários
Hormônio Secretado Tipo de Adenoma Síndrome Clínica Frequência Relativa
Prolactina Prolactinoma Hipogonadismo,
galactorreia
25-40%
GH Somatotrófico Acromegalia/gigantismo 10-15%
ACTH Corticotrófico Doença de Cushing 10-15%
LH/FSH Gonadotrófico Pan-Hipopituitarismo 10-15%
TSH Tireotrófico Hipertireoidismo < 3%
Nenhum Não funcionante Pan-hipopituitarismo 10-25%
c) Hipofisite
• É a inflamação da hipófise, a qual pode ocorrer por diversas causas.
• Deve ser desconfiada principalmente nos seguintes pacientes:
o Histórico de doença endócrina autoimune + hipopituitarismo
o Mulheres com aumento do tamanho da hipófise e hipopituitarismo durante gestação ou período
pós-parto.
d) Síndrome de Sheehan
• É uma causa clássica de hipopituitarismo.
• Só pra lembrar: é típico no pós-parto, em que o parto foi complicado por alterações hemodinâmicas
significantes, levando a necrose hipofisária por hipofluxo sanguíneo. Mais de 75% da adeno-hipófise
deve ser acometida.
• Geralmente as pacientes têm agalactia (falta de prolactina), amenorreia secundária (falta de LH e
FSH) e sintomas inespecíficos (adinamia, astenia, letargia, anorexia – fruto da baixa de outros
hormônios). Devido a falta do ADH, as pacientes podem se apresentar com
poliúria/polidipsia/noctúria, sintomas de diabetes insipidus, porém, essa é uma manifestação rara.
3) Quadro Clínico
• O desenvolvimento de sinais e sintomas costuma ser lento e insidioso, dependendo do início e da
magnitude da lesão hipotálamo-hipofisária.
• Geralmente, a sintomatologia é inespecífica e pode se manifestar com um quadro de fraqueza, mal-estar,
letargia, frio, perda de peso, perda de apetite, dor abdominal, entre outras queixas.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Frequentemente, quando a causa do hipopituitarismo é adquirida, os sintomas e déficts hormonais
surgem na seguinte ordem: GH → LH, FSH → TSH → ACTH → PRL (exceto em casos de hipofisite,
quando o ACTH costuma ser o primeiro eixo acometido)
• No caso de lesões com efeito de massa, podem estar presentes sintomas compressivos, como alterações
visuais e neurológicas.
o A cefaleia é um sintoma comum, podendo ser retro-orbitária ou referida para a parte superior do
crânio. Seu início súbito associado a outros sintomas (náuseas, vômitos) indica apoplexia.
o O quiasma óptico é a estrutura geralmente acometida, sendo que é a estrutura responsável por
conduzir as fibras provenientes dos dois campos temporais da visão. Portanto, o
acometimento dessa estrutura leva a hemianopsia bitemporal (perda de visão lateral – o
paciente irá frequentemente esbarrar em objetos com a parte lateral do corpo)
o A compressão dos nervos cranianos adjacentes (III, IV, VI e V) pode levar aos seguintes
sintomas:
▪ III Nervo: perda do reflexo fotomotor e consensual (midríase paralítica)
▪ III, IV e/ou VI nervo: oftalmoplegia com estrabismo
▪ V nervo: perda do reflexo corneopalpebral
• ATENÇÃO! Diferentemente da IA primária, a zona glomerular e o SRAA estão íntegros no
hipopituitarismo, portanto, não se observa desidratação, depleção de sódio ou hiperpotassemia
• Ao exame físico, geralmente os pacientes se encontram um pouco acima do peso normal. A pele
costuma ser fina, pálida e lisa, com finas rugas na face. A pilificação pode estar deficiente e ocorrer
hipotensão postural. Redução de força muscular e hiporreflexia podem estar presentes.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
4) Diagnóstico
• A avaliação diagnóstica do paciente com suspeita de hipopituitarismo deve determinar:
o A presença, o tipo e o grau de deficiência hormonal
o A etiologia da deficiência hormonal
o A presença de alterações visuais
• Abaixo, alguns achados laboratoriais dos principais diagnósticos
o Deficiência de GH
▪ GH basal, IGF-1 e IGF-BP3 baixos ou normais
▪ Durante ITT, pico de GH < 5 ng/ml em crianças e < 3 ng/ml em adultos
o Deficiencia de ACTH
▪ ACTH basal: baixo ou normal baixo (< 10 pg/ml)
▪ Cortisol Basal < 3 ug/dl
▪ Durante ITT, pico de cortisol < 18 mg/dl
o Deificência de TSH
▪ TSH basal: baixo ou normal, às vezes, algo elevado (TSH biologicamente inativo)
Sintomas de acordo com o hormônio acometido
GH FSH/LH ou
GnRH
TSH ACTH ou
CRH
PRL
- Período neonatal
• Hipoglicemia
• Icterícia
• Hipotermia
• Micropênis
• ↓Crescimento
- Crianças
• ↓Crescimento
• Voz fina
• Ganho de
peso (lipólise
reduzida)
• Alteração na
dentição
- Adultos
• ↓Bem estar
• Astenia
• Ganho de
peso
• Depressão
• Labilidade
emocional
• Resistencia à
insulina
• Dislipidemia
• Hipertensão
diastólica
- Período neonatal
• Criptoquidia
• Micropênis
- Crianças
• Atraso no
desenvolvimento
puberal
• Atraso na idade
óssea
- Mulheres
• Amenorreia
• ↓Libido
• Infertilidade
• Secura vaginal
• Dispaureunia
• Osteoporose
• Sintomas
climatéricos
- Homens
• Disfunção erétil
• Hipotrofia
testicular
• Redução da
libido
• Infertilidade
• Redução da
massa muscular
e óssea
• Depressão
• Défict
cognitivo
• Intolerancia
ao frio
• Ganho de
peso
• Rouqidão
• Edema
• Sonolencia
• Constipação
intestinal
• Perda de peso
• Anorexia
• Fraqueza
• Náusea
• Dor abdominal
• Febre
• Choque
(Situações de
estresse –
ausência de
glicocorticoides
para
reatividade
vascular)
• Perda da
lactação

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
▪ T4 livre baixo, T3 normal ou baixo
o Deficiencia de gonadotrofinas
▪ LH e FSH basais: baixos ou normais
▪ Testosterona baixa (em homens) e estradiol baixo (em mulheres)
• ATENÇÃO! Geralmente, para o diagnóstico da deficiência de GH e ACTH, tornam-se necessários os
testes de estímulo, como o teste de tolerância à insulina (ITT).
o ITT: coleta-se amostras para dosagem de cortisol após administração IV de insulina regular.
Apresenta como efeitos adversos sudorese, palpitação, tremor e crise convulsiva. É
contraindicado em idosos, portadores de doenças vasculares, cardiopatas e pessoas com
histórico de convulsão.
o Estímulo com ACTH: a hipófise, por se encontrar atrofiada, nem sempre responde a esse
estímulo. São raros os efeitos colaterais e não apresenta contra-indicações.
o L-arginina: utilizada para dosagem de GH (infunde-se arginina e mensura-se o GH). Seu
principal efeito colateral são náuseas.
o L-arginina + GHRH: mesma coisa do teste acima. Seu principal efeito colateral é rubor.
5) Tratamento
• Os hormônios hipofisários apresentam secreção pulsátil e ritmos secretórios que variam com uma
enorme gama de fatores. Por isso, não é possível a reposição fisiológica dos hormônios hipofisários
para o tratamento do hipopituitarismo, o qual é feito quase sempre por meio da substituição dos
hormônios dos órgãos alvo.
a) Deficiência de GH
• Os níveis de IGF-1 devem ser utilizados como marcador de tratamento, devendo ser mantidos entre o
valor médio e o limite superior da faixa de referência ajustada para a idade do paciente, visando a um
nível ótimo de função física e psicossocial.
• A reposição hormonal é feita com GH humano recombinante (rhGH), administrado por via
subcutânea antes de dormir.
• Efeitos colaterais: cefaleia, artralgia, mialgia, síndrome do túnel do carpo, edema de extremidades
(retenção hídrica provocada pelo GH)
• Contraindicações: doença malgina ativa, hipertensão intracraniana benigna e retinopatia diabética
proliferativa.
• ATENÇÃO! A reposição não está indicada e não há qualquer benefício demonstrado em estados de
deficiência funcional de GH, como observado no processo de envelhecimento (“somatopausa”),
obesidade e síndrome metabólica.
• ATENÇÃO! Nas deficiências hormonais, o ideal é começar com a reposição de rhGH apenas quando os
outros déficts já estejam adequadamente tratados. É importante monitorar os níveis séricos de cortisol

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
e T4 livre, pois quadros subclinicos de ACTH e TSH podem se tornar manifestos com o início do
tratamento do rhGH.
b) Deficiência de ACTH (insuficiência adrenal secundária ou terciária)
• Há necessidade apenas de repor os glicocorticoides, pois o SRAA não é afetado na IA por déficit de
ACTH.
• O objetivo do tratamento é repor, de modo mais fisiológico possível, a menor dose de glicocorticoide
que mantenha o paciente livre dos sintomas de insuficiência adrenal e sem os riscos de hipercortisolismo
iatrogênico (continua sendo um risco com o tratamento prolongado)
• Reposição mais utilizada: hidrocortisona VO na dose de 20 mg (10 mg ao acordar, 5 mg ao meio-dia e
5 mg ao fim da tarde). Em situações de estresse (cirurgia, doença), o paciente deve dobrar ou triplicar a
dose do GC oral ou fazer reposição por via parenteral.
• Crise Adrenal (quadro agudo): hidrocortisona IV, 50 a 100 mg
• Quadros crônicos e assintomáticos: reposição de GC em situações de estresse, orientando o paciente a
carregar consigo um cartão/bracelete em caso de crise adrenal.
• ATENÇÃO! As doses do GC podem necessitar de ajuste em pacientes que iniciam tratamento com
rhGH, já que o GH inibe a conversão de cortisona em cortisol. Os ajustes de dose dependem da
avaliação clínica, pois não há parâmetro bioquímico e hormonal para avaliação de resposta.
• ATENÇÃO! A reposição de deidroepiandrosterona em mulheres para correção da insuficiência
androgênica não é recomendada.
c) Deficiência de TSH (hipotireoidismo central)
• É idêntico ao tratamento do primário, e consiste na administração de levotiroxina (T4 livre) em dose
única ao acordar pela manhã, antes de se alimentar.
• A dose inicial deve variar entre 50 e 100 ug/dia (exceto em idosos e cardiopatas), com ajustes até se
atingir a dose de manutenção (50 a 200 ug/dia). Essa dose deve ser um pouco maior nos pacientes em
uso concomitante de estrógenos e rhGH.
• ATENÇÃO! Com o início do tratamento com rhGH ou estrogênios, os níveis séricos de T4 livre devem
ser monitorados para eventuais ajustes da dose de tiroxina. Uma queda nos níveis de T4 livre é esperada,
visto que o GH aumenta a conversão de T4 para T3.
• ATENÇÃO! A presença de hipocortisolismo deve ser tratada antes ou durante a reposição do
hormônio tireoidiano, pois o T4 livre pode aumentar a necessidade de glicocorticoides, eventualmente
precipitando uma crise adrenal.
d) Deficiência de FSH e LH (hipogonadismo hipogonadotrófico)
• O tratamento do hipogonadismo, obviamente, difere entre homens e mulheres.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
1. Homens
• Reposição de testosterona, a qual pode ser feita por via transdérmica, intramuscular, intranasal, bucal,
ou na forma de implantes subcutâneos.
• Alguns estudos tem mostrado reversibilidade do hipogonadismo hipogonadotrófico congênito em cerca
de 5 a 10% dos pacientes com reposição androgênica, portanto, é válida a tentativa de suspensão
periódica dessa reposição para verificar possibilidade de interrupção definitiva do tratamento.
• Quando o objetivo for promover aumento testicular ou restauração da fertilidade, pode-se empregar
GnRH pulsátil ou terapia combinada com gonadotrofinas.
• A reposição de esteroides gonadais por 3 a 4 meses é uma abordagem usada em casos de retardo puberal,
sendo muitas vezes útil para diferenciar o hipogonadismo hipogonadotrófico do atraso constitucional da
puberdade.
2. Mulheres
• Deve ser feita a reposição estroprogesterônica, sendo que várias preparações estão disponíveis. Deve-se
dar preferência aos estrogênios naturais para o tratamento a longo prazo, sobretudo ao estradiol e ao
17B-estradiol.
• Sobre as vias de administração: uma importante desvantagem da via oral em relação ao uso em gel ou
transdérmico é a primeira passagem hepática, que resulta em síntese de proteínas pró-inflamatórias e
pró-trombóticas, além de ter efeitos indesejáveis sobre a pressão arterial e os níveis de SHBG (em
contrapartida, apresenta melhora do perfil lipídico). O Vilar orienta a dar preferência às preparações
estrogênicas transdérmicas em pacientes que necessitem reposição de GH; estrogenoterapia oral
implica em doses muitos mais altas de rhGH.
• O tratamento de infertilidade dessas pacientes apresenta riscos e é extremamente complexo, devendo ser
realizado somente em centros de reprodução com experiência em técnicas de estimulação ovariana.
• ATENÇÃO! A reposição estrogênica em mulheres com útero deve sempre ser acompanhada da
administração cíclica ou contínua de progestógenos, visando minimizar o risco de hiperplasia ou câncer
do endométrio, bem como promover maior regularidade nos ciclos menstruais.
3) Descrever as principais causas de tumores hipofisários funcionantes: prolactinoma, acromegalia e
Doença de Cushing
1) Prolactinoma
a) Introdução
• A hiperprolactinemia se caracteriza por níveis séricos elevados de prolactina, cuja principal função é
estimular a lactação. Apesar de sua prevalência baixa na população em geral, sua prevalência é mais
elevada em indivíduos com sintomas relacionados ao excesso da secreção desse hormônio, como
amenorreia, galactorreia e infertilidade.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• A hiperprolactinemia em si é apenas um achado laboratorial, o qual pode resultar de causa fisiológicas,
farmacológicas e patológicas. A principal etiologia patológica são os prolactinomas, adenomas
secretores de prolactina.
o Fisiológicas: gravidez e amamentação, estresse, exercício, manipulação da mama e sono.
o Farmacológicas: antipsicóticos (antagonizam receptores D2 na hipófise anterior) e
antidepressivos são os mais relacionados a esse quadro (risperidona, fenotiazinas e
bunofenonas).
o Patológicas:
▪ Tumores: prolactinoma e tumores que cursam com produção aumentada de PRL ou
comprometimento da haste hipotálamo-hipofisária,
▪ Hipotireoidismo: o aumento do TRH estimula a síntese de PRL e o lactotrofo tem
sensibilidade reduzida ao efeito da dopamina e da queda de T3 e T4
▪ Doença de Addison: os glicocorticoides suprimem a expressão do gene da PRL e sua
liberação
▪ Cirrose Hepática
▪ Insuficiência Renal (redução da depuração renal da PRL – principalmente em
pacientes com hemodiálise)
▪ Hiperprolactinemia neurogênica: elevação reflexa de PRL em decorrência de lesões
irritativas da parede torácica (herpes-zóster, toracotomia, queimaduras) e por
patologias do cordão medular (ependimoma cervical, siringomielia, tumore
extrínsecos).
▪ Crise convulsiva: convulsão do lobo frontal e temporal pode levar a desequilíbrio dos
neurotransmissores da região hipotálamo-hipofisária.
o Macroprolactinemia: condição suspeitada quando o paciente se apresenta sem sintomas típicos
ou evidência de tumores a RM. Está associada a menor biodisponibilidade da prolactina.
• Os prolactinomas são a principal causa patológica de hiperprolactinemia. Na prática clínica,
microprolactinomas (< 1 cm) são mais prevalentes do que macroprolactinomas (> 1 cm) e ocorrem mais
frequentemente em mulheres com idade entre 20 e 50 anos (pode estar relacionado ao subdiagnóstico em
homens, devido ao quadro clínico mais típico em mulheres – síndrome de amenorreia-galactorreia).
o Em homens, os macroadenomas são mais frequentes.
b) Quadro Clínico
• O quadro clínico se dá pelo excesso de prolactina, cursando com os seguintes sintomas:
o Galactorreia: pode ser espontânea, intermitente ou detectável apenas à expressão mamilar. Pode
estar ausente em casos graves de hipogonadismo (como pós-menopausa). Em homens, é
praticamente patognomônica de prolactinoma.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
o Hipogonadismo: causado pela inibição da secreção do GnRH hipotalâmico por parte da
prolactina.
▪ Em mulheres manifesta-se por anovulação, infertilidade, oligo ou amenorreia e
redução da lubrificação vaginal com dispaureunia.
▪ Em homens pode cursar com redução da libido, disfunção erétil, oligospermia,
infertilidade, ejaculação precoce e, menos frequentemente, ginecomastia.
▪ Em ambos os sexos cursa com redução da densidade mineral óssea.
o Outras manifestações: hirsutismo e acne, obesidade e alterações devido a expansão tumoral.
▪ A expansão tumoral cursa com os seguintes sintomas, a depender de sua direção:
• Infrasselar: rinorreia liquórica, com risco de meningite
• Suprasselar: cefaleia, redução ou perda de visão e hemianopsia bitemporal,
hipertensão craniana ou hidrocefaleia (compressão do III ventrículo).
• Parasselar: oftalmoplegia e/ou dor facial
c) Diagnóstico
• O diagnóstico é feito pela constatação de hiperprolactinemia associado a achados de imagem
sugestivos do tumor (TC ou RM). Sempre lembrar de excluir possíveis causas fisiológicas e
farmacológicas para o quadro.
o Hiperprolactinemia: PRL de 20 a 25 ng/ml em mulheres e 15 a 20 ng/ml em homens. Em casos
duvidosos, é de bom grado pedir outra amostra (elevações discretas da PRL podem ser fruto da
pulsatilidade da secreção do hormônio ou estresse da venopunção)
▪ ATENÇÃO! Níveis séricos > 250 ng/ml são quase patognomônicos dos
prolactinoma. Níveis > 500 ng/ml são capazes de confirmar esse diagnóstico.
▪ ATENÇÃO! Os níveis de PRL podem se encontrar falsamente baixos em volumosos
macroprolactinomas e acentuada hiperprolactinemia, devido ao efeito gancho
(saturação de anticorpos de captura e sinalizadores no imunoensaio).
▪ Em geral, é possível correlacionar os níveis de prolactina e o tamanho do tumor.
• Microprolactinomas: PRL entre 100 e 250 ng/ml
• Macroprolactinomas: PRL entre 250 e 1.000 ng/ml, podendo exceder esse
valor, principalmente nos casos de prolactinomas gigantes.
o Exames de imagem: úteis para detectar o tumor e classifica-lo em microadenoma (diâmetro <
1 cm), macroadenoma (diâmetro > 1cm) ou prolactinomas gigantes (> 4 cm). Os principais
são a TC e a RM.
• ATENÇÃO! O principal diagnóstico diferencial são os pseudoprolactinomas (adenomas clinicamente
não funcionantes), os quais cursam com elevação dos níveis de prolactina devido a compressão da haste
hipofisária, sendo o seu tratamento cirúrgico.
o Nessa condição, os valores de PRL costumam ser < 100 ng/ml

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• ATENÇÃO! Outro diagnóstico diferencial importante é a macroprolactinemia, situação em que há
predomínio da macroprolactina, levando a uma mensuração laboratorial elevada de PRL, contudo sem
repercussões clínicas (visto que a molécula é biologicamente inativa). Pode cursar com sintomatologia,
quando associada a distúrbios concomitantes (hiperprolactinemia farmacológica, síndrome dos ovários
policísticos...)
d) Tratamento
• O tratamento dos prolactinomas pode ser realizado por meio de cirurgia, radioterapia e farmacoterapia
com agonistas dopaminérgicos (esse último é o de escolha para a situação). Pacientes assintomáticos
com microprolactinomas não apresentam indicação absoluta de tratamento (deve ser instituído
apenas caso surjam sintomas).
• Cirurgia: indicada principalmente para os casos de
resistência ou intolerância ao tratamento farmacológico,
assim como nos que existem complicações tumorais
(apoplexia ou rinoliquiorreia). Em casos de
prolactinomas gigantes e invasivos, dificilmente a
cirurgia pode ser curativa. A técnica de escolha costuma
ser a cirurgia transesfenoidal, que apresenta como
intercorrência comum o diabetes insípido transitório
• Radioterapia: indicada apenas para tumores resistentes a
DA e cirurgia (como massa tumoral significativa ou que
cresça durante o seguimento), com tendência
comprovada para crescimento. Os prolactinomas são
tumores hipofisários secretores menos responsivos à
radioterapia. A complicação crônica mais frequente é o
hipopituitarismo induzido por radiação (mais de 50%
dos pacientes que receberem radioterapia desenvolverão
no mínimo uma deficiência de hormônio da hipófise
anterior na década seguinte).
• Farmacológico: é feita através de agonistas dopaminérgicos, sendo o tratamento de primeira linha
para esses tumores (bromocriptina e cabergolina). Seu mecanismo de ação se dá pela estimulação dos
receptores dopaminérgicos D2, responsáveis por inibir e reduzir a síntese e secreção de prolactina.
o Carbergolina: é o agonista mais utilizado para tratamento, devido a sua seletividade para o
receptor D2. Sua posologia também é útil: só necessita de uma única dose oral apenas a ½ vezes
por semana, enquanto a bromocriptina requer três tomadas diárias. Além disso, estudos
demonstram que apresenta efeito metabólico benéfico, sendo capaz de reduzir a prevalência de
síndrome metabólica e resistência insulínica nesses pacientes. É o tto de escolha, pois, além dos

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
benefícios supracitados, se mostrou mais eficaz em normalizar PRL e induzir redução tumoral e
menor taxa de intolerância do que a bromocriptina.
o Bromocriptina: fármaco de segunda escolha, geralmente é administrada 2 a 3 vezes/dia. Pode
causar hipotensão postural e, em doses elevadas, fenômeno de Raynauld. Usada na gestação.
o Efeitos colaterais: náuseas, vômitos, cefaleia, tontura, congestão nasal, dor abdominal,
fenômeno de Raynauld. Depressão, psicose, rinoliquorreia e herniação do quiasma óptico são
efeitos colaterais mais raros, porém presentes. Alguns estudos indicam risco de valvopatia
(regurgitação tricúspide) devido a presença de receptores dopaminérgicos nas valvas cardíacas
(leva a mitogenese e proliferação de fibroblastos) → ainda está em discussão a necessidade
de realizar ecocardiograma de screening (apenas devem ser acompanhados pacientes que
utilizam doses elevadas por períodos prolongados).
• ATENÇÃO! A grande maioria dos tumores da hipófise são lesões benignas, de crescimento lento e não
invasivas. Entretanto, podem estar presentes tumores com invasão local, recidiva após cirurgia e
resistência a tratamento medicamentoso. Esses se enquadram como carcinomas (<1% dos tumores
hipofisários) ou adenomas atípicos. O prognóstico desses pacientes é reservado, porém estudos
documentam o uso de temozolomida como eficaz para tumores agressivos produtores de PRL ou ACTH
(como é um fármaco que induz à mielosupressão, deve ser utilizado apenas em último caso).
2) Acromegalia
a) Introdução
• A acromegalia é uma doença sistêmica crônica, decorrente da produção excessiva de GH e IGF-1.
• Em quase 100% dos casos, a acromegalia é causada por um adenoma hipofisário secretor de GH
(somatotropinoma). Esse adenoma pode ser de células puras de GH ou misto, com células de GH e
prolactina.
o Apesar de raras, existem outras causas de acromegalia, como: adenomas mistos de células
secretoras de GH e prolactina, adenomas ectópicos secretores de GH, carcinoma de células
somatotróficas, adenomas pluri-hormonais e linfomas.
• É uma doença rara, porém certamente é subdiagnosticada.
• Acomete igualmente homens e mulheres, principalmente entre 30 e 50 anos.
o Em crianças se manifesta por crescimento linear excessivo e gigantismo (ocorre antes do
fechamento das cartilagens de crescimento).
• Em razão de sua evolução insidiosa, seu diagnóstico é frequentemente feito em torno de 7 a 10 anos após
o aparecimento dos primeiros sinais e sintomas. Isso é importante pelo fato de o diagnóstico e o
tratamento mais precoces poderem evitar ou minimizar o surgimento das complicações cardiovasculares
respiratórias e neoplásicas, principais responsáveis pelo aumento de mortalidade nessa condição.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
b) Quadro Clínico
• É um distúrbio insidioso, com progressão gradual dos sinais e sintomas, os quais podem resultar da
compressão do tumor sob estruturas adjacentes, da secreção excessiva de GH ou das complicações
sistêmicas resultantes do excesso desse hormônio.
• Há aumento das extremidades em praticamente 100% dos casos, mas não é motivo de consulta por
parte dos pacientes, que atribuem ao ganho de peso o fato de anéis, luvas e sapatos ficarem mais
apertados.
• Em crianças, pode estar presente o gigantismo, devido ao aumento da secreção excessiva de GH antes
do fechamento das cartilagens de crescimento. Deve ser considerado em crianças que estejam mais de
três desvios padrões acima da altura média para a idade ou mais de dois DP além da altura ajustada para
a altura dos pais.
• Modificações fisionômicas: alargamento do nariz, aumento dos lábios, crescimento exagerado da
mandíbula com prognatismo, proeminência frontal, separação dos dentes, maloclusão dentária,
macroglossia e aumento dos arcos zigomáticos. Devido ao caráter insidioso de seu surgimento, são o
motivo de consulta inicial de apenas 10% dos pacientes.
• Manifestações articulares: artralgia (70% dos casos) e artropatia
• Manifestações cutâneas: hiperidrose, pele oleosa com odor desagradável, espessamento cutâneo. Os
acrocórdons (skin tags) podem ser observados em alguns pacientes, sendo importantes marcadores para
a existência de pólipos colônicos adenomatosos. O fenômeno de Raynaud pode ser observado em até um
terço dos acromegálcios.
• ATENÇÃO! Frequentemente o diagnóstico é estabelecido casualmente, a partir de manifestações
clínicas frutos de complicações da doença, como irregularidades menstruais, diabetes melito, apneia do
sono, síndrome do túnel do carpo...
Complicações da acromegalia
Cardiovasculares Respiratórias Endócrinas Metabólicas Neoplasias
- Hipertensão
- Cardiomegalia
- Hipertrofia
ventricular
- Arritmias
- AVC
- Apneia do
sono
- Depressão de
centros
respiratórios
- Calcificação
traqueal
- Artropatia da
junta
cricoaritenóidea
- Hiperprolactinemia
- Hipopituitarismo
- Anormalidade
menstruais
- Disfunção erétil
- Diabetes Melito
- Hipertrigliciridemia
- Hipercalcemia
- Hipercalciúra
- Tireoide
- Pólipos
- Cólon
- Esôfago
- Mama
- Estômago
- Tireoide
c) Diagnóstico
• Envolve a avaliação de aspectos clínicos, laboratoriais e radiológicos.
• A suspeita clínica da doença é confirmada a partir da dosagem basal de GH e IGF-1.
o Exames adicionais são RM da sela túrcica e exames de imagem toracoabdominais.
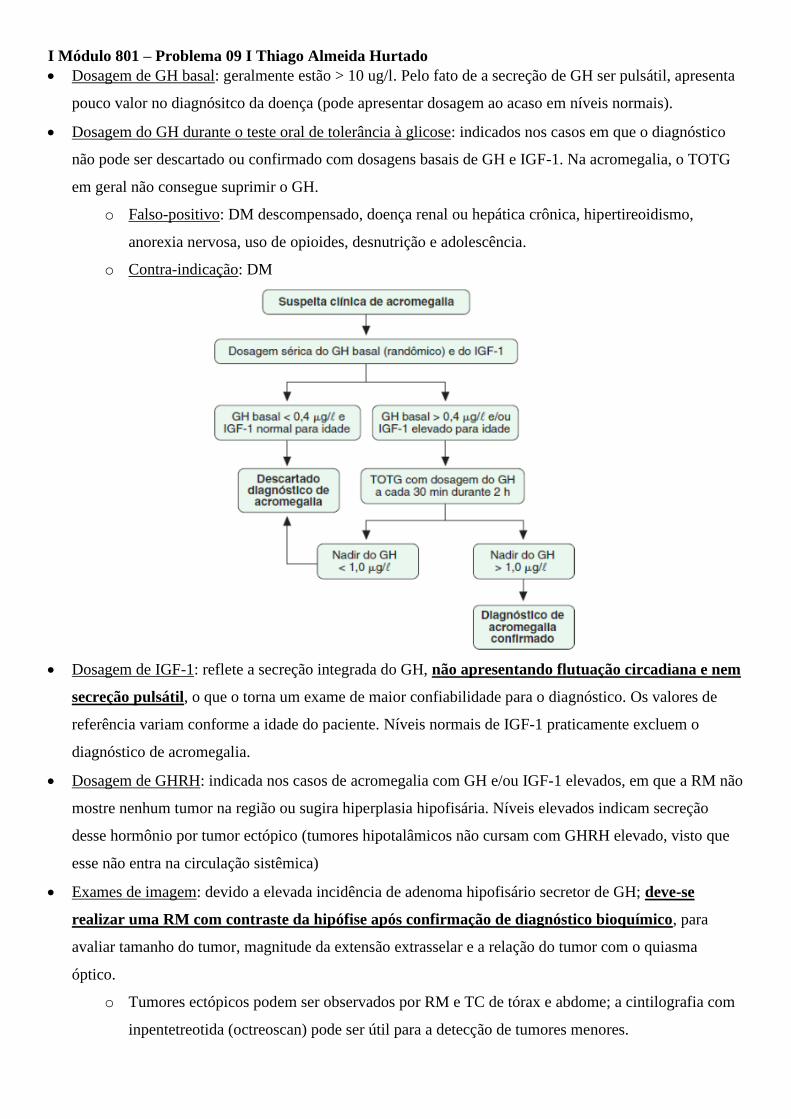
I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Dosagem de GH basal: geralmente estão > 10 ug/l. Pelo fato de a secreção de GH ser pulsátil, apresenta
pouco valor no diagnósitco da doença (pode apresentar dosagem ao acaso em níveis normais).
• Dosagem do GH durante o teste oral de tolerância à glicose: indicados nos casos em que o diagnóstico
não pode ser descartado ou confirmado com dosagens basais de GH e IGF-1. Na acromegalia, o TOTG
em geral não consegue suprimir o GH.
o Falso-positivo: DM descompensado, doença renal ou hepática crônica, hipertireoidismo,
anorexia nervosa, uso de opioides, desnutrição e adolescência.
o Contra-indicação: DM
• Dosagem de IGF-1: reflete a secreção integrada do GH, não apresentando flutuação circadiana e nem
secreção pulsátil, o que o torna um exame de maior confiabilidade para o diagnóstico. Os valores de
referência variam conforme a idade do paciente. Níveis normais de IGF-1 praticamente excluem o
diagnóstico de acromegalia.
• Dosagem de GHRH: indicada nos casos de acromegalia com GH e/ou IGF-1 elevados, em que a RM não
mostre nenhum tumor na região ou sugira hiperplasia hipofisária. Níveis elevados indicam secreção
desse hormônio por tumor ectópico (tumores hipotalâmicos não cursam com GHRH elevado, visto que
esse não entra na circulação sistêmica)
• Exames de imagem: devido a elevada incidência de adenoma hipofisário secretor de GH; deve-se
realizar uma RM com contraste da hipófise após confirmação de diagnóstico bioquímico, para
avaliar tamanho do tumor, magnitude da extensão extrasselar e a relação do tumor com o quiasma
óptico.
o Tumores ectópicos podem ser observados por RM e TC de tórax e abdome; a cintilografia com
inpentetreotida (octreoscan) pode ser útil para a detecção de tumores menores.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Diagnósticos diferenciais: DM descompensado, cirrose, anorexia nervosa, uremia, doenças agudas,
SIDA, desnutrição... São condições que cursam com produção ou ação deficiente do GH e suas proteínas
carreadoras. Manifestações observadas no hipotireoidismo grave podem lembrar as da acromegalia.
Em crianças, o gigantismo pode estar relacionado a outras condições, como alta estatura familiar,
síndrome de Marfan e hiperinsulinismo.
d) Tratamento
• São três as modalidades terapêuticas: cirurgia, tratamento medicamentoso e radioterapia.
• Cirurgia: único tratamento que tem chance de cura imediata do quadro (eficácia de 90% em
microadenomas e de 50% nos macroadenomas); é considerada a terapia de primeira escolha na
maioria dos casos. Além disso, fornece tecido tumoral para análise histopatológica e molecular, o que
permite melhor caracterização do tumor e tratamento adjuvante mais apropriado na ausência de cura.
Quando realizada por neurocirurgiões experientes, o índice de complicações é baixíssimo (a mais
frequente é o diabetes insípido transitório). O hipopituitarismo e a recorrência da acromegalia são
outras complicações.
• Farmacológico: devem ser considerados para esse os pacientes com níveis séricos de IGF-1 normais
para a idade e GH randômico < 1,0 ug/l. Os fármacos utilizados são análogos da somatostatina,
agonistas dopaminérgicos e antagonistas do receptor do GH.
o Análogos da somastotatina: são o octreotide e o lanreotide. São indicados como terapêutica
adjuvante à cirurgia ou como tratamento primário naqueles tumores com baixa chance de
cura cirúrgica (extrasselares). Sua aplicação é intramuscular ou subcutânea e os principais
efeitos colaterais são gastrintestinais.
o Agonistas dopaminérgicos: são a carbergolina e a bromocriptina. Assim como nos
prolactinomas, a carbegolina apresenta melhores resultados que a bromocriptina. É
preferencialmente utilizada em adenomas cossecretores de GH e prolactina. Pode ser
utilizado como monoterapia adjuvante em pacientes com níveis pouco elevados de GH e IGF-1.
Seus principais efeitos adversos já foram descritos.
o Antagonistas do Receptor de GH: é o pegvisomanto (PEG-V); indicada para pacientes com
resistência ou intolerância aos análogos da somatostatina, ou naqueles em que houve falência
do tratamento cirúrgico. Agem se ligando aos receptores hepáticos e bloqueando a produção de
IGF-1. Não apresenta efeito sobre a massa tumoral (ação periférica), a qual pode inclusive
aumentar. Elevações de transaminases e lipo-hipertrofia são os efeitos adversos mais comuns.
• Radioterapia: é uma modalidade eficaz no controle da doença, contudo, devido ao risco de efeitos
colaterais graves, é utilizada apenas para tumores não controlados com tratamento cirúrgico e
medicamentoso. O hipopituitarismo é o efeito mais frequente, entretanto, lesão de vias ópticas ou de

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
pares cranianos, déficit cognitivo, doença cerebrovascular e malignidades secundárias também podem
aparecer.
3) Doença de Cushing ACTH dependente
OBS pros calouros: essa parte também está no problema 06, mas o resumo de lá foi feito pelo Medcurso
2019, e esse daqui pelo livro da Patrícia.
a) Introdução
• A síndrome de Cushing é um estado clínico resultante do excesso de cortisol circulante do organismo
(decorrente da ingestão de glicocorticoide ou da produção endógena)
• As etiologias dividem-se entre ACTH-dependente (80% dos casos) e ACTH independente (15 a
20%). A primeira apresenta como principal etiologia o adenoma de hipófise, já a segunda apresenta
como principal etiologia o adenoma adrenal.
o ACTH-dependente: adenoma de hipófise (doença de Cushing); tumor produtor de ACTH
ectópcio, síndrome do CRH ectópico
o ACTH-independennte: adenoma adrenal, carcinoma adrenal, hiperplasias da adrenal
• É uma condição rara, com incidência de 0,7 a 2,4 por 1.000.000 de habitantes por ano. É mais comum
em mulheres.
b) Quadro Clínico
• Pode causar um largo espectro de manifestações, desde quadros subclínicos até quadros mais
exuberantes, a depender da duração e intensidade de produção dos glicocorticoides.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Obesidade centrípeta e visceral: principalmente na fossa supraclavicular e no tronco, fácies em lua cheia
e giba cervical. Pode desencadear síndrome da apneia obstrutiva do sono por deposição acentuada de
gordura em região cervical.
• Manifestações cutâneas: estrias violáceas largas (> 1cm), equimoses espontâneas (fragilidade capilar por
redução da síntese de colágeno), escurecimento da pele nos casos de ACTH dependente (devido a
produção de melanotrofina derivada da POMC). Hirsutismo, alopecia, hiperandrogenismo e acne
podem estar presentes devido a produção de andrógenos. A acantose nigricans pode estar presente
devido a resistência insulínica (cortisol é hiperglicemiante)
• Sistema musculoesquelético: miopatia e fraqueza proximal (glicocorticoide é catabólico), osteoporose
(redução na reabsorção intestinal e renal de cálcio – pode ser agravada pelo hipogonadismo)
• Sistema cardiovascular: hipertensão arterial sistêmica (aumento do angiotensinogênio, dano vascular,
vasoconstrição sistêmica e perda do descenso noturno da pressão dependente do ciclo do cortisol),
dislipidemia (ativação da lipase lipoproteica subcutânea, aumentando a lipólise e liberando AGL e TG),
hipercoagulabilidade/estado pró-trombótico.
o A dislipidemia apresenta-se com hipercolesterolemia e hipertrigliceridemia. O fármaco de
escolha deve ser a rouvastatina/pravastatina, pois a sinvastatina apresenta metabolismo reduzido
com o uso de cetoconazol, o que aumenta o risco de toxicidade medicamentosa.
• Sistema neurológico: agitação, ansiedade, depressão, pânico e alterações cognitivas e de memória
• Hipogonadismo hipogonadotrófico: amenorreia, perda de libido, disfunção erétil e infertilidade (cortisol
inibe GnRH).
• Infecções de repetição: onicomicose ou tínea versicolor.
• Pediatria: atraso da idade óssea e déficit do crescimento.
c) Diagnóstico
• Sempre questionar acerca de fontes exógenas de corticoide (nasal, cremes, comprimidos)
1. Confirmação do hipercortisolismo
• Cortisol livre urinário: deve ser feito em urina de 24h. Pode ser falso-positivo nos casos de poliúria, uso
de glicocorticoides e síndrome pseudo-cushing. Pode ser falso-negativo quando a coleta é inadequada e
em casos de insuficiência renal.
• Cortisol salivar à meia noite: devem ser coletadas preferencialmente duas amostras. Orientar o paciente a
não beber nem fumar por 24 horas, não comer 1h antes do exame, não escovar os dentes 2h antes e lavar
a boca com água antes de coletar o exame. Pode ser falso-positivo (sangue na amostra, idosos, estresse,
trabalhadores noturnos)
• Cortisol sérico à meia noite: deve ser coletado com o paciente internado (de preferência após 48h de
internação para reduzir o estresse dessa).

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Teste de supressão com dose baixa de dexametasona: inibe a secreção de ACTH pela adeno-hipófise.
Pode ser feito através da administração de 1mg de dexametasona (coleta no dia seguinte) ou pelo
protocolo de Liddle 1 (0,5 mg de dexametasona de 6/6h durante 48h com coleta de cortisol sérico na
manhã do terceiro dia).
2. Distinção entre formas ACTH-dependente ou independente
• Deve ser confirmado o hipercortisolismo com pelo menos dois dos testes anteriormente citados.
• É feito através da dosagem do ACTH
o ACTH < 5 pg/ml → ACTH independente
o ACTH 5 a 20 pg/ml → necessidade de realização de testes dinâmicos (caso responsivo é ACTH
dependente)
o ACTH > 20 pg/ml → ACTH dependente
3. Testes dinâmicos
• Teste de desmopressina (DDAVP): realizado para diferenciar doença de Cushing de tumor ectópico
secretor de ACTH. Coleta-se o ACTH e o cortisol sérico após a administração de desmopressina. Um
aumento de 35% no ACTH e de 20% no cortisol sugerem Doença de Cushing. A resposta “aumentada”
da Doença de Cushing se deve a presença de mais receptores para esse hormônio nos adenomas
corticotróficos, induzindo a uma maior secreção de ACTH.
• Teste do CRH humano ou ovino: realizado para diferenciar doença de Cushing de tumores ectópicos
coleta-se o ACTH e o cortisol sérico após a administração de CRH humano ou ovino IV. Um incremento
de 105% de ACTH e de 14% de cortisol (quando administrado CRH humano) sugerem Doença de
Cushing (adenomas corticotróficos são mais ricos em receptores de CRH). É muito mais sensível e
específico do que o DDAVP, porém é mais caro e menos disponível.
• Teste de supressão com dose alta de dexametasona ou Liddle 2: Administra-se dexametasona 2mg Vo
6/6h ou dexametasona 8 mg à meia-noite. Dosa-se o cortisol sérico pela manhã ou urinário 24h do dia
seguinte da última dose. Altas doses de glicocorticoides suprimem parcialmente a secreção de ACTH em
adenomas corticotróficos, ao contrário do que ocorre em tumores adrenais e na secreção ectópica de
ACTH.
4. Averiguar a etiologia/localização
• RM hipofisária: imagens > 6 mm sugerem o diagnóstico de doença de Cushing. Se a imagem for menor
do que 6 mm, realizar cateterismo bilateral para confirmar a hipótese. 40 a 50% dos casos podem não ter
imagem visível na RM e cerca de 10% da população pode ter incidentalomas hipofisários (geralmente <
6mm).
• Cateterismo bilateral e simultâneo dos seios petrosos inferiores: é o método padrão ouro para
diferenciação entre fonte hipofisária e não hipofisária de síndrome de Cushing ACTH-dependente.
Cateterizam-se os seios petrosos e uma veia periférica, mensurando-se o gradiente de ACTH após

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
administração com CRH ou DDAVP. Um gradiente centro/periferia > 2 (basal) ou > 3 (pós-estímulo
com secretagogo) sugere doença de Cushing.
• Exames localizatórios em caso de suspeita de tumor ectópico: TC de tórax, abdome, pelve e marcadores
tumorais.
d) Tratamento
• O padrão ouro sempre será a cirurgia, idealmente por via transesfeinoidal. Caso haja falha, pode ser
indicada nova cirurgia ou tratamento medicamentoso, adrenalectomia uni ou bilateral ou radioterapia.
o ATENÇÃO! Ao contrário do tto medicamentoso na acromegalia e nos prolactinomas, na DC
os medicamentos não são capazes de reduzir o tamanho tumoral

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado
• Cirurgia hipofisária: tto de primeira linha, geralmente realizado por via transesfeinoidal (90% dos casos
são microadenomas). Para os casos que não entram em remissão, deve ser realizada uma segunda
cirurgia.
• Traatmento medicamentoso: indicado para pacientes sem controle cirúrgico ou enquanto aguardam os
efeitos tardios da radioterapia. Melhora as comorbidades do paciente, portanto, pode também ser
utilizada no pré e pós-operatório e enquanto o diagnóstico de hipercortisolismo ACTH-dependente não
estiver firmamente estabelecido.
1. Moduladores da secreção de ACTH
• Cabergolina (agonista dopaminérgico): geralmente utilizada em associação com outros tratamentos,
como o cetoconazol (melhora a resposta, pois o hipercortisolismo reduz a expressão de receptores D2
nos corticotrofos).
2. Medicamentos com ação nas adrenais
• Cetoconazol: é o medicamento mais utilizado para o controle do hipercortisolismo. Apresenta um
mecanismo de ação triplo: inibe as enzimas da esteroidogênese adrenal e gonadal, reduz a produção de
ACTH e apresenta ação competitiva com o cortisol nos receptores de glicocorticoides.
o Efeitos colaterais: elevação das enzimas hepáticas, intolerância TGI, cefaleia, sedação,
ginecomastia, redução de libido, disfunção erétil.
• Mitotane: apresenta efeito citotóxico sobre as adrenais, inibindo a esteroidogênese e apresentando efeito
adrenolítico direto. Seu início de ação é demorado (~6 semanas), porém, apresenta efeito prolongado.
o Efeitos colaterais: insuficiência adrenal (dose-dependente), intolerância de TGI,
hepatotoxicidade, ginecomastia, artralgia, leucopenia, dislipidemia, rash, teratogenicidade.
• Etomidato: medicamento IV, usado em casos graves, que inibe a secreção aguda de cortisol.
• Adrenalectomia videolaparoscópica: indicada apenas em pacientes com falha no tratamento cirúrgico
que não podem ou não desejam passar por uma nova abordagem neurocirúrgica. Apresenta certeza de
cura quando bilateral, às custas de insuficiência adrenal para o resto da vida.
• Radioterapia: indicada apenas nos casos sem cura cirúrgica e sem melhor alternativa terapêutica.
Apresenta início de ação lento, demorando cerca de 12 a 18 meses para cursar com melhora do
hipercortisolismo, além de poder evoluir com hipopituitarismo à longo prazo. Deve ser sempre associada
a algum tipo de tratamento medicamentoso devido a seu efeito demorado.

I Módulo 801 – Problema 09 I Thiago Almeida Hurtado


















