
DIAGNÓSTICO DAS DHM: RASTREIO NEONATAL … · estado nas dietas hipoproteicas 1992 1ºs passos...
24
DIAGNÓSTICO DAS DHM: RASTREIO NEONATAL ALARGADO XIV CURSO BÁSICO DOENÇAS HEREDITÁRIAS METABOLISMO 13/12/2017 Casa Acreditar - Coimbra
Transcript of DIAGNÓSTICO DAS DHM: RASTREIO NEONATAL … · estado nas dietas hipoproteicas 1992 1ºs passos...

DIAGNÓSTICO DAS DHM: RASTREIO NEONATAL ALARGADO
XIV CURSO BÁSICO DOENÇAS HEREDITÁRIAS METABOLISMO
13/12/2017
Casa Acreditar - Coimbra

SUMÁRIO
- História
- O Presente
Organização
Colheitas
Laboratório
Resultados
Tratamento
Doenças Rastreadas – revisão
- Panorama Mundial
- Considerações Finais

UM POUCO DE HISTÓRIA
1979
Rastreio da
Fenilcetonúria
1981
Hipotiroidismo
Congénito
Fenilcetonúria
90s
Estudos piloto:
- Hiperplasia Congénita da
Supra-Renal
- Deficiência em Biotinidase
- Fibrose Quística (FQ)
Hipotiroidismo Congénito
Fenilcetonúria
≥2004
Estudo piloto:
- Outras Doenças Hereditárias do
Metabolismo (DHM)
Hipotiroidismo Congénito
Fenilcetonúria
1987
Comparticipação do
estado nas dietas
hipoproteicas 1992
1ºs passos para a APOFEN
2002
Espetrómetros de massa

UM POUCO DE HISTÓRIA
2008
Rastreio
sistemático a
nível nacional:
25 patologias
2013
Reinicio do rastreio
da Fibrose Quística
25 patologias
2016
Diário da República
Despacho nº 3653/2016
Centros de Referência Nacionais
para o Tratamento das Doenças
Hereditárias do Metabolismo
NGS
Fibrose Quística
25 patologias
2017
Diário da República
Despacho nº 6669/2017
Centros de Referência Nacionais
na área da Fibrose Quística
Fibrose Quística
25 patologias
+ Centros de Referência
2015
Rastreio da
Drepanocitose
países africanos Marcadores de 2ºnível
Técnica HPLC-MS/MS isómeros C5-carnitina
Doseamento da homocisteína total
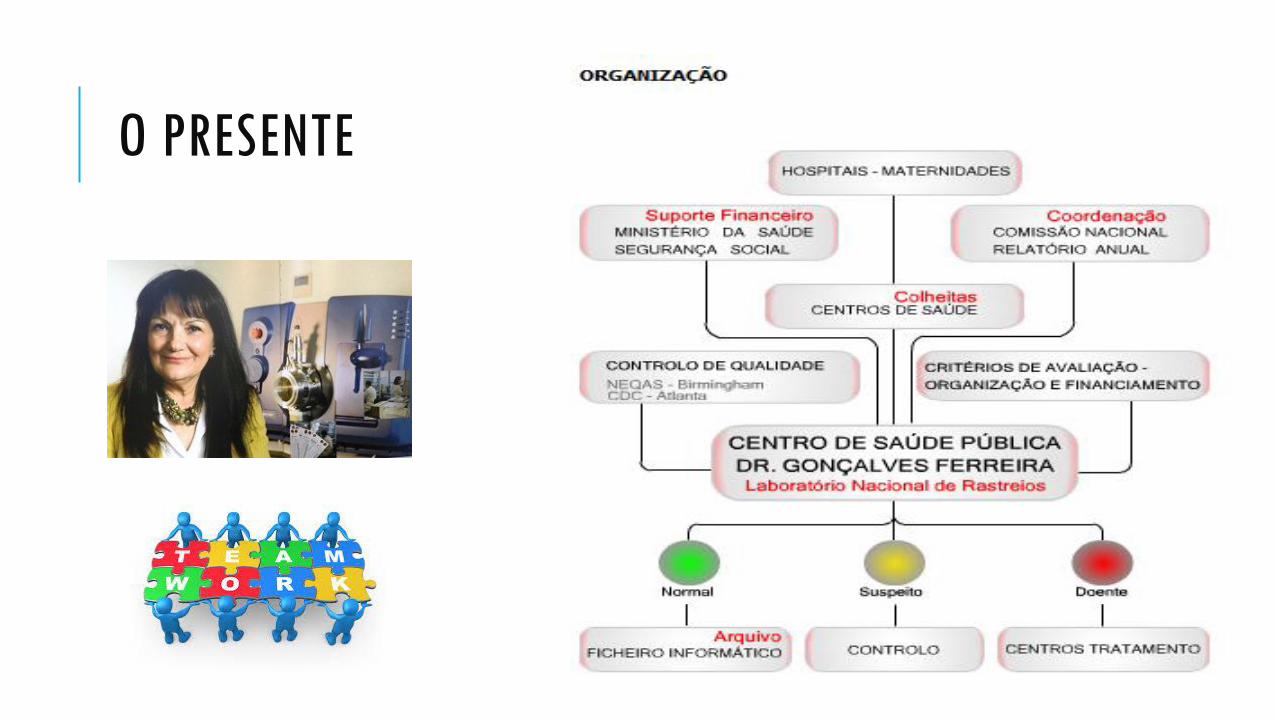
O PRESENTE

O PRESENTE - COLHEITAS
3º- 6ºdias de vida
Cuidados de Saúde 1ª > 2º e 3º
Ou de uma veia aquando de colheitas para outros fins

O PRESENTE - COLHEITAS CASOS ESPECIAIS
Pré-termo
- < 30 SG e/ou peso <1500 g
- 3 colheitas
- 3º - 6º dias
- após 2 semanas (14º - 15º dias)
- 4 semanas
Localidades
- Baixo Alentejo: etnia cigana leucinose - colheita antes da alta da maternidade

O PRESENTE - COLHEITAS
Fonte: PNDP Relatório 2016
Número de recém-nascidos estudados e número de nascimentos, por ano Idade do recém-nascido na altura da colheita, por ano
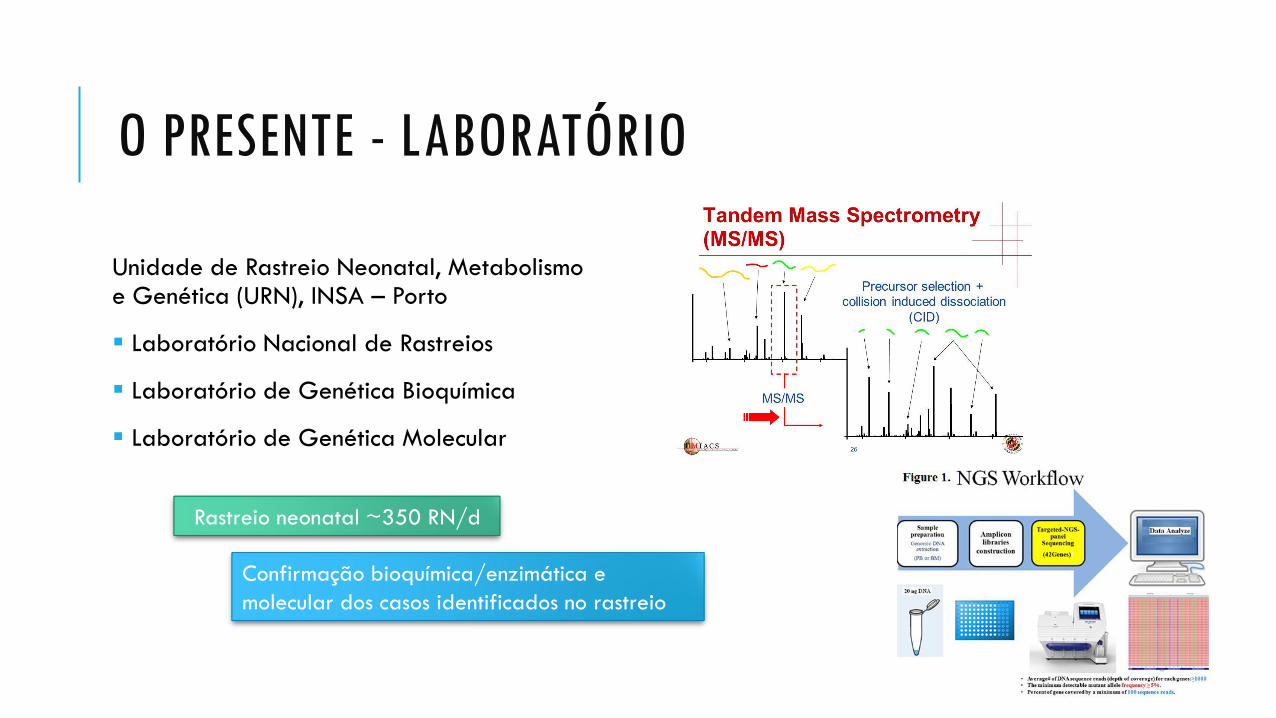
O PRESENTE - LABORATÓRIO
Unidade de Rastreio Neonatal, Metabolismo e Genética (URN), INSA – Porto
Laboratório Nacional de Rastreios
Laboratório de Genética Bioquímica
Laboratório de Genética Molecular
Rastreio neonatal ~350 RN/d
Confirmação bioquímica/enzimática e
molecular dos casos identificados no rastreio
O PRESENTE - RESULTADOS
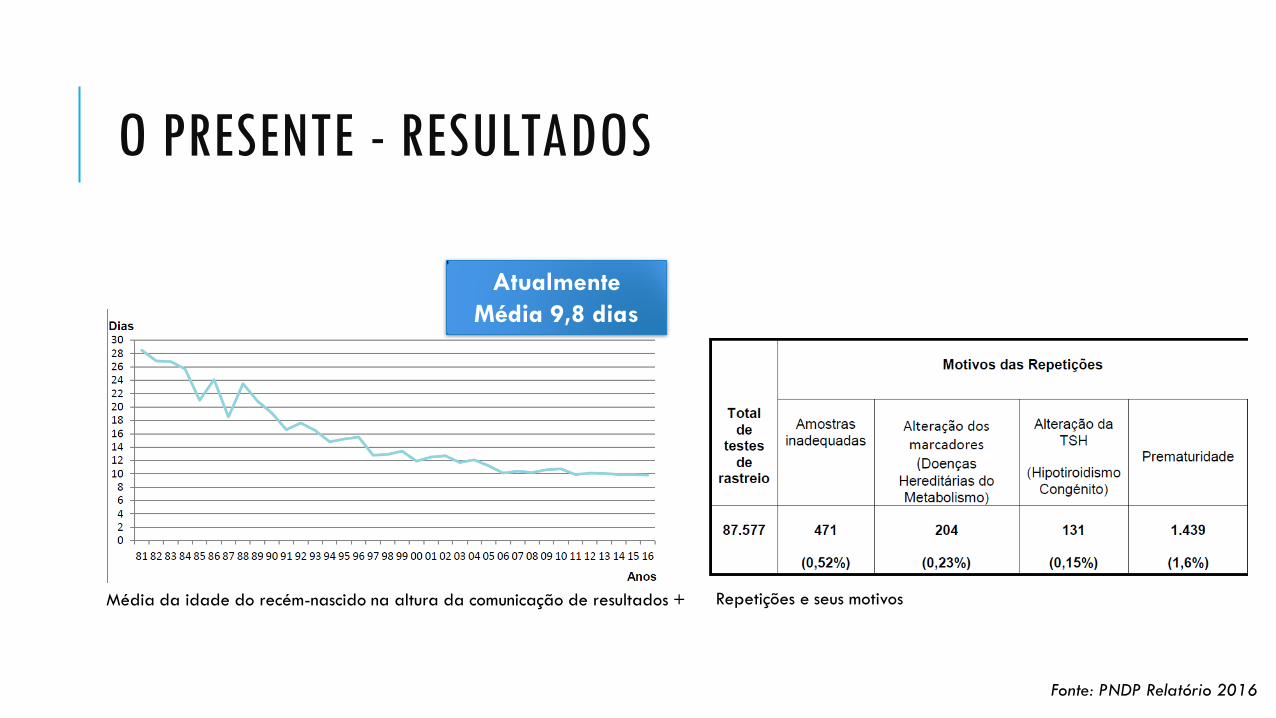
O PRESENTE - RESULTADOS
Média da idade do recém-nascido na altura da comunicação de resultados +
Atualmente
Média 9,8 dias
Fonte: PNDP Relatório 2016
Repetições e seus motivos

O PRESENTE - RESULTADOS
Falsos positivos existem!
Em 2016:
défice de cobalamina mãe vegan, RN sob aleitamento materno exclusivo
carnitinas baixas doença materna
Também se pode chegar ao diagnóstico de outras DHM!
tirosina e metionina elevadas citopatia mitocondrial

O PRESENTE - TRATAMENTO
Centros de Referência DHM – 2016 e FQ – 2017
Centro Hospitalar do Porto, E. P. E.
Centro Hospitalar de São João, E. P. E.
Centro Hospitalar Universitário de Coimbra, E. P. E.
Centro Hospitalar Lisboa Norte, E. P. E.
Centro Hospitalar Lisboa Central, E. P. E.
Consulta de Nutrição do Centro Hospitalar do Porto
responsável nacional pela gestão da distribuição dos produtos hipoproteicos
comparticipação do Estado, Despacho nº14319/2015, de 29 de junho

O PRESENTE - DOENÇAS RASTREADAS
Hipotiroidismo Congénito
Fibrose Quística
Doenças Hereditárias do Metabolismo
Aminoacidopatias
Acidúrias Orgânicas
Doenças Hereditárias da ß-oxidação Mitocondrial dos Ácidos Gordos
Incidência 1:2136
39 RN
diagnosticados
4 mães
diagnosticadas
Incidência 1:2246
Incidência 1:7735

DHM RASTREADAS
Aminoacidopatias
Fenilcetonúria (PKU) / Hiperfenilalaninemias
Tirosinemia Tipo I
Tirosinemia Tipo II
Leucinose (MSUD)
Citrulinemia Tipo I
Acidúria Arginino-Succínica
Hiperargininemia
Homocistinúria Clássica
Hipermetioninemia (Déf. MAT)
Fonte: Research Gate
Perfil de aminoácidos de um doente com fenilcetonúria
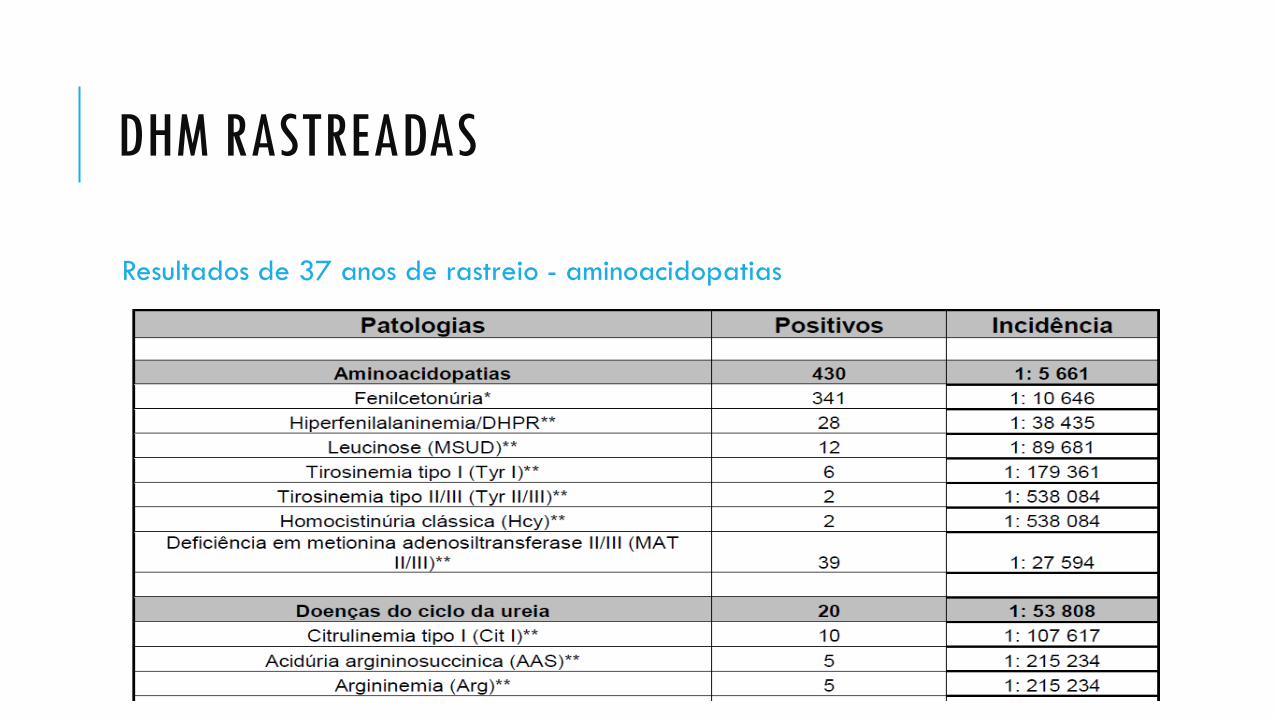
DHM RASTREADAS
Resultados de 37 anos de rastreio - aminoacidopatias

DHM RASTREADAS
Acidúrias Orgânicas
Acidúria Propiónica (PA)
Acidúria Metilmalónica (MMA, Mut-)
Acidúria Isovalérica (IVA)
Acidúria 3-Hidroxi-3-Metilglutárica
(3-HMG)
Acidúria Glutárica Tipo I (GA I)
3-Metilcrotonilglicinúria (Déf. 3-MCC)
Acidúria Malónica
Fonte: Atlas of Inherited Metabolic Diseases
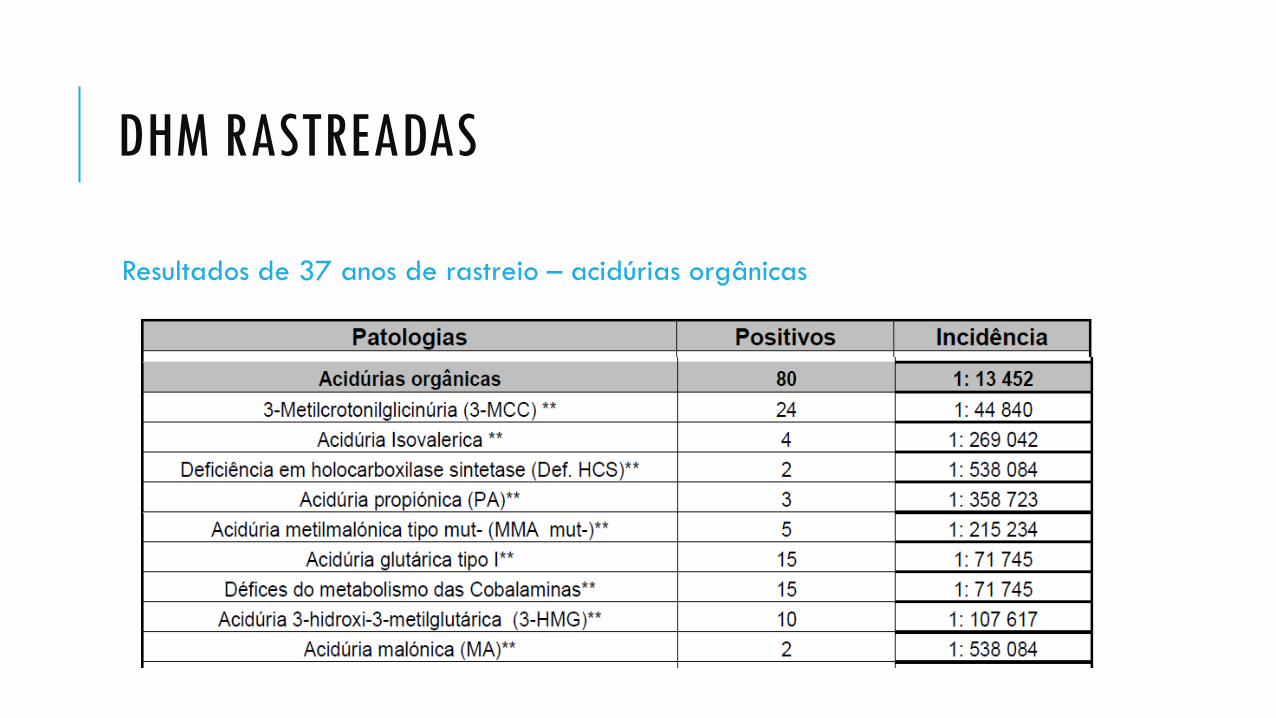
DHM RASTREADAS
Resultados de 37 anos de rastreio – acidúrias orgânicas
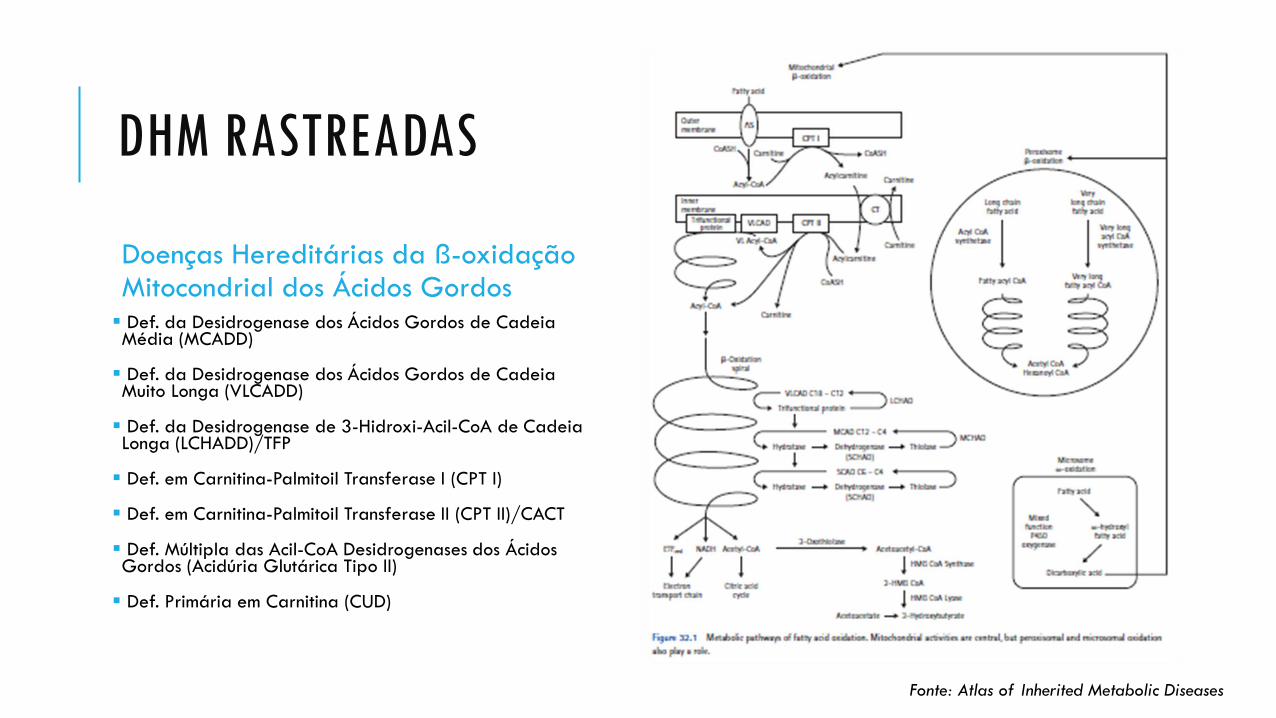
DHM RASTREADAS
Doenças Hereditárias da ß-oxidação Mitocondrial dos Ácidos Gordos Def. da Desidrogenase dos Ácidos Gordos de Cadeia Média (MCADD)
Def. da Desidrogenase dos Ácidos Gordos de Cadeia Muito Longa (VLCADD)
Def. da Desidrogenase de 3-Hidroxi-Acil-CoA de Cadeia Longa (LCHADD)/TFP
Def. em Carnitina-Palmitoil Transferase I (CPT I)
Def. em Carnitina-Palmitoil Transferase II (CPT II)/CACT
Def. Múltipla das Acil-CoA Desidrogenases dos Ácidos Gordos (Acidúria Glutárica Tipo II)
Def. Primária em Carnitina (CUD)
Fonte: Atlas of Inherited Metabolic Diseases
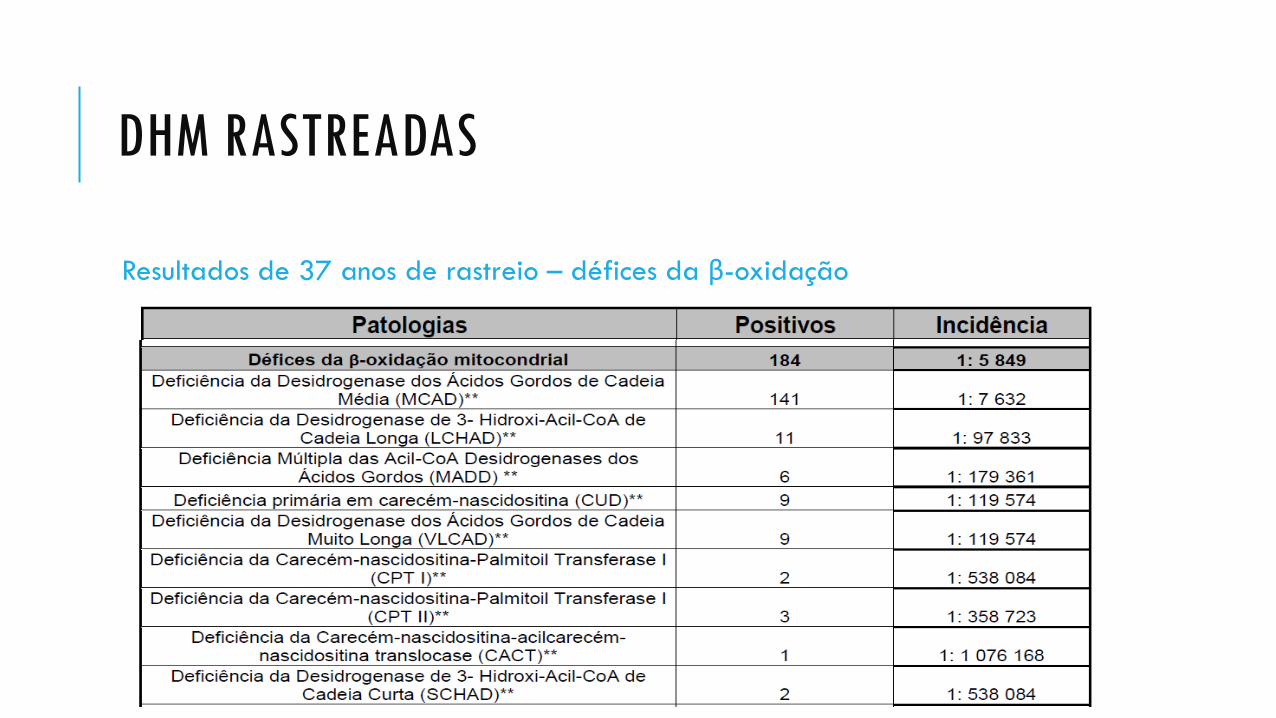
DHM RASTREADAS
Resultados de 37 anos de rastreio – défices da β-oxidação

PANORAMA MUNDIAL

CONSIDERAÇÕES
Programa Nacional com uma cobertura de 100%
Bem aceite e acarinhado pela população
Objetivo: antecipar/prevenir descompensações melhorar o prognóstico
Deteção em fase pré-sintomática:
famílias menos cientes da gravidade da doença
Existem falsos positivos
Casos que eventualmente nunca serão sintomáticos

CONSIDERAÇÕES
Trabalho contínuo
Avaliação de doenças como potenciais novas integrantes do PNDP
Avaliação da justificação da manutenção das que já o integram
Avaliação da aplicabilidade das novas técnicas
Journal of Inherited Metabolic Disease
March 2017, Volume 40, Issue 2, pp 209–218
Long-term outcome of expanded newborn screening at Boston children’s hospital: benefits and challenges in defining true disease

OBRIGADA PELA ATENÇÃO!


















