
UNIVERSIDADE FEDERAL DE PERNAMBUCO - UFPE · inovação terapêutica tem tomado proporções...
137
UNIVERSIDADE FEDERAL DE PERNAMBUCO Centro de Ciências Biológicas Programa de Pós-Graduação em Inovação Terapêutica Luiz Felipe Gomes Rebello Ferreira Desenvolvimento e implementação de software para aplicação de grids computacionais em modelagem para inovação terapêutica Recife 2013
Transcript of UNIVERSIDADE FEDERAL DE PERNAMBUCO - UFPE · inovação terapêutica tem tomado proporções...

UNIVERSIDADE FEDERAL DE PERNAMBUCO
Centro de Ciências Biológicas
Programa de Pós-Graduação em Inovação Terapêutica
Luiz Felipe Gomes Rebello Ferreira
Desenvolvimento e implementação de software para aplicação
de grids computacionais em modelagem para inovação
terapêutica
Recife
2013

Luiz Felipe Gomes Rebello Ferreira
Desenvolvimento e implementação de software para
aplicação de grids computacionais em modelagem para
inovação terapêutica
Dissertação de Mestrado apresentada ao
Programa de Pós-Graduação em Inovação
Terapêutica da Universidade Federal de
Pernambuco, para a obtenção do Título de
Mestre em Inovação Terapêutica
Orientador: Prof. Dr. Marcelo Zaldini Hernandes
Recife
2013

Catalogação na fonte Elaine Barroso
CRB 1728
Ferreira, Luiz Felipe Gomes Rebello Desenvolvimento e implementação de software para aplicação de grids computacionais em modelagem para inovação terapêutica/ Luiz Felipe Gomes Rebello Ferreira– Recife: O Autor, 2013. 122 folhas : il., fig., tab.
Orientador: Marcelo Zaldini Hernandes Dissertação (mestrado) – Universidade Federal de
Pernambuco, Centro de Ciências Biológicas, Inovação Terapêutica, 2013. Inclui bibliografia e apêndices
1. Bioinformática 2. Grid computacional 3. Moléculas I.
Hernandes, Marcelo Zaldini (orientador) II. Título 570.28 CDD (22.ed.) UFPE/CCB- 2013- 064

UNIVERSIDADE FEDERAL DE PERNAMBUCO
CENTRO DE CIÊNCIAS BIOLÓGICAS
PROGRAMA DE PÓS-GRADUAÇÃO EM INOVAÇÃO TERAPÊUTICA
Recife, 21 de fevereiro de 2013.
Dissertação de Mestrado defendida e APROVADA, por decisão unânime, em 21 de
fevereiro de 2013, cuja Banca Examinadora foi constituída pelos seguintes professores:
PRESIDENTE E PRIMEIRO EXAMINADOR INTERNO: Prof. Dr. Marcelo
Zaldini Hernandes
(Departamento de Ciências Farmacêuticas da Universidade Federal de Pernambuco)
Assinatura:_______________________________________
SEGUNDO EXAMINADOR INTERNO: Prof. Dr. César Augusto Souza de
Andrade
(Departamento de Bioquímica da Universidade Federal de Pernambuco)
Assinatura:_______________________________________
PRIMEIRO EXAMINADOR EXTERNO: Prof Dr Carlos Henrique Madeiros
Castelletti
(Instituto Agronômico de Pernambuco)
Assinatura:_______________________________________

DEDICATÓRIA
Dedico esta dissertação a minha família.

AGRADECIMENTOS
Ao orientador Marcelo Zaldini pelo apoio, paciência e inúmeras
sugestões dadas para esta dissertação.
Ao Klaus Cavalcante por ter me iniciado nesta área, pelo apoio nos
momentos mais difíceis e por toda ajuda e sugestões dadas ao longo
destes últimos anos.
A minha família pelo apoio incondicional.
Aos amigos do Laboratório de Química Medicinal (LQTM).
Ao programa de pós-graduação em inovação terapêutica (PPGIT).
A Fundação de Amparo à Ciência e Tecnologia do Estado de
Pernambuco (FACEPE) pelo auxilio financeiro a este trabalho através da
concessão da bolsa de estudos.

RESUMO
A utilização de computadores no desenvolvimento de produtos ligados à
inovação terapêutica tem tomado proporções significativas, particularmente na
área de planejamento molecular baseado em métodos computacionais. Estima-
se que o uso destas metodologias pode reduzir os custos e o tempo de
desenvolvimento de um novo fármaco em até 50%. Isto ocorre porque muitas
vezes o número de moléculas que precisam ser sintetizadas e testadas
experimentalmente passa a ser drasticamente reduzido por conta da alta
preditividade e confiabilidade dos métodos computacionais (in silico). O docking
molecular determina se pode haver interação energética favorável entre duas
moléculas (ligante e alvo biológico), no intuito de elucidar as razões
moleculares responsáveis pela potência farmacológica destes fármacos em
potencial. Ocorre que estes métodos podem apresentar, por vezes, uma alta
demanda computacional quando o número de ligantes e alvos a serem
testados é alto e quando se busca alta precisão nos resultados numéricos
obtidos. Este trabalho apresenta a plataforma GriDoMol para execução de
cálculos de docking molecular no ambiente distribuído através de um grid
computacional. O programa GriDoMol pode ser usado tanto com o programa
AutoDock quanto com o programa AutoDock Vina, para realizar os cálculos de
docking. Através da interface intuitiva do GriDoMol é possível acompanhar o
andamento com dados do conjunto de cálculos de docking e criar o arquivo Job
Description File (JDF) contendo a lista de cálculos de docking que será
realizado em paralelo no ambiente de grid computacional. Tanto no programa
AutoDock quanto no AutoDock Vina, o tempo necessário para a realização de
500 cálculos de docking molecular foi reduzido em até 97% do tempo
necessário quando comparado a não utilizar nenhuma estratégia de
paralelização, ou de computação distribuída.
Palavras-chave: GriDoMol. grid computacional. docking molecular.
computação distribuída. inovação terapêutica.

ABSTRACT
The use of computers in developing products related to therapeutic
innovation has taken significant proportions, particularly in the area of
computer-based molecular design. It is estimated that the use of these methods
can reduce the costs and time spent at development stage of a new drug by
50%. This often happens because the number of molecules that need to be
experimentally synthesized and tested becomes drastically reduced due to the
high predictability and reliability of the computational methods (in silico). The
molecular docking procedure determines if there is favorable interaction energy
between two molecules (ligand and biological target) in order to elucidate the
molecular reasons responsible for the pharmacological potency of these
potential drugs. It happens that these methods may present, sometimes, a high
computational demand when the number of ligands and targets to be tested is
high and when seeking for a high precision in the numerical results. This work
presents the GriDoMol platform for performing molecular docking calculations in
a distributed system through a computational grid environment. GriDoMol can
be used either with the program AutoDock as the program AutoDock Vina, to
perform molecular docking calculations. Through the GriDoMol intuitive
interface, one can track the progress of the docking calculations and create the
Job Description File (JDF) containing the list of docking calculations to be
performed in parallel at the grid computing environment. Both in AutoDock and
in AutoDock Vina, the time required to perform 500 tasks of molecular docking
was reduced by up to 97% of the time required when compared to not using any
parallelism strategy, or distributed computing.
Keywords: GriDoMol. computational grid. molecular docking. distributed
computing.

LISTA DE ILUSTRAÇÕES
Figura 2.1 – Exemplo de um complexo estrutural (PDB: 1ULB) formado por um receptor (cor
laranja) e seu ligante co-cristalizado (modelo de esferas e ligações, em cores azul e branca). O
melhor resultado de re-docking molecular pode ser observado detalhadamente (modelo de
ligações, em cor vermelha), bem como sua alta superposição com a estrutura do ligante co-
cristalizado. (a) A visão panoramica da superfice do receptor, incluindo o ligante co-cristalizado
e a solução de docking. (b) Uma visão detalhada do sitio ativo do receptor.............................. 06
Figura 2.2 – Heterogeneidade e distribuição geográfica de um grid computacional.................. 17
Figura 5.1 – Tela inicial do GriDoMol, sendo executado no sistema operacional Windows....... 47
Figura 5.2 – Tela do GriDoMol para criação do job no Windows................................................ 48
Figura 5.3 - Esquema da arquitetura de comunicação entre os programas de cada máquina
utilizados neste trabalho.............................................................................................................. 52
Figura 5.4 - Gráfico de desempenho do AutoDock em função do tempo. As curvas com a
legenda no formato Ex (onde x se refere ao número de núcleos de processamento utilizados)
se refere aos valores observados enquanto as curvas no formato Exp representam os valores
previstos...................................................................................................................................... 56
Figura 5.5 - Gráfico do FdI para o programa AutoDock. As legenda no formato Mx (onde x se
refere ao número de máquinas utilizadas) mostra os valores observados em relação ao
previsto........................................................................................................................................ 58
Figura 5.6 – Gráfico do FdM para o programa AutoDock. As legenda no formato Ex (onde x se
refere ao número de instâncias de execução utilizadas) mostra os valores observados em
relação ao previsto...................................................................................................................... 59
Figura 5.7 – Gráfico de dispersão dos cálculos individuais de docking com o programa
AutoDock ao utilizar: (a) uma máquina e uma instância de execução; (b) uma máquina e oito
instâncias de execução; (c) seis máquinas e uma instância de execução................................. 68
Figura 5.8 – Gráfico do desempenho do AutoDock Vina em função do tempo. As curvas com a
legenda no formato Nx (onde x se refere ao número de núcleos de processamento utilizados)
se refere aos valores observados enquanto as curvas no formato Nxp representam os valores
previstos...................................................................................................................................... 71
Figura 5.9 – Gráfico dos valores de FdC no programa AutoDock Vina. A legenda no formato Mx
(onde x se refere ao número de máquinas utilizadas) se refere aos valores observados.......... 72
Figura 5.10 – Gráfico dos valores de FdM no programa AutoDock Vina. A legenda no formato
Mx (onde x se refere ao número de máquinas utilizadas) se refere aos valores observados.... 73
Figura 5.11 – Gráfico de dispersão dos cálculos individuais de docking com o programa
AutoDock Vina ao utilizar: (a) uma máquina e um núcleo de processamento; (b) uma máquina
e oito núcleos de processamento; (c) oito máquinas e um núcleo de processamento cada...... 81
Figura 5.12 – Fator de aceleração do desempenho, a nivel de núcleos de processamento dos
cálculos individuais de docking no AutoDock Vina, ao utilizar os oito núcleos de processamento
em relação a utilizar apenas um núcleo de processamento. A linha vermelha representa o valor
médio de aceleração do desempenho........................................................................................ 83

Figura 5.13 – Desempenho ao reordenar a sequência dos cálculos individuais de docking. O
eixo das ordenadas representa o tempo em segundos, enquanto o eixo das abscissas
representa as 5 repetições de cada teste................................................................................... 86
Figura 5.14 – Gráficos do desempenho dos 10 ordenamentos aleatórios. O M1C8 se refere a
uma máquina utilizando oito núcleos de processamento enquanto o M8C8 a oito máquinas
utilizando oito núcleos de processamento...................................................................................88

LISTA DE TABELAS
Tabela 1 – Comparativo entre alguns pontos chaves entre cluster e grid computacional.......... 13
Tabela 2 – Lista do conjunto de complexos ligante-receptor selecionados para este estudo.... 41
Tabela 3 – Características do código fonte implementado no programa GriDoMol obtido a partir
do programa SLOCCount............................................................................................................ 46
Tabela 4 – Total de cálculos realizados...................................................................................... 53
Tabela 5 – Desempenho computacional comparativo do GriDoMol durante a realização dos
cálculos com o AutoDock............................................................................................................ 55
Tabela 6 – Tempo total estimado de comunicação entre os componentes do ambiente de grid
computacional............................................................................................................................. 61
Tabela 7 – Tempos de execução individual de cada cálculo de docking, no AutoDock, ao utilizar
uma máquina e uma instância do programa AutoDock por máquina. As colunas E1 a E10
representa o tempo de execução individual de cada cópia de cada complexo dentro do mesmo
job................................................................................................................................................ 65
Tabela 8 – Tempos de execução individual de cada cálculo de docking, no AutoDock, ao utilizar
uma máquina e oito instâncias simultâneas do programa AutoDock por máquina. As colunas E1
a E10 representa o tempo de execução individual de cada cópia de cada complexo dentro do
mesmo job................................................................................................................................... 66
Tabela 9 – Tempos de execução individual de cada cálculo de docking, no AutoDock, ao utilizar
seis máquinas e apenas uma instância do programa AutoDock por máquina. As colunas E1 a
E10 representa o tempo de execução individual de cada cópia de cada complexo dentro do
mesmo job................................................................................................................................... 67
Tabela 10 – Desempenho computacional comparativo do GriDoMol durante a realização dos
cálculos com o AutoDock Vina.................................................................................................... 70
Tabela 11 – Cálculos refeitos para elucidar os valores elevados de FdM encontrados em alguns
dos cálculos................................................................................................................................. 74
Tabela 12 – Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar uma máquina e um núcleo de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job............. 78
Tabela 13 – Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar uma máquina e oito núcleos de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job............. 79

Tabela 14 – Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar oito máquina e um núcleo de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job............. 80
Tabela 15 – Tempos de execução dos cálculos utilizando diferentes sequências de
ordenamento das tarefas individuais (docking)........................................................................... 85
Tabela 16 – Tempos de execução dos jobs aleatórios, em segundos....................................... 87
LISTA DE ABREVIAÇÕES E SIGLAS
BAT Batch (Arquivo de Lote)
BoT Bag of tasks (Conjunto de tarefas)
CADD Computer Aided Drug Design (Desenvolvimento de fármacos com o auxílio de
métodos computacionais)
CCB Centro de Ciências Biológicas
CNPq Conselho Nacional de Desenvolvimento Científico e Tecnológico
COCOMO Construtive Cost Model (Modelo de estimativa de tempo de desenvolvimento)
CPU Central Processing Unit (Unidade central de processamento)
DLG Docking Log File (Arquivo de registro de docking)
DPF Docking Parameters File (Arquivo de parâmetros de docking)
FACEPE Fundação de Amparo à Ciência e Tecnologia do Estado de Pernambuco
FdC Fator de divisão por Cores (núcleos de processamento)
FdI Fator de divisão por Instâncias de execução
FdM Fator de divisão por Máquinas
GPF Grid Parameters File (Arquivo de parâmetros do AutoGrid)
GPU Graphics Processing Unit (Unidade de processamento gráfico)
GSF GriDoMol Setup File (Arquivo de configurações de docking do GriDoMol)
HIV Human immunodeficiency vírus (Vírus da imunodeficiência humana)
IDE Integrated Development Environment (ambiente de desenvolvimento integrado)
JDF Job Description File (Arquivo de descrição das tarefas)
LQTM Laboratório de Química Teórica Medicinal
MOL2 Arquivo de estrutura molecular desenvolvido pela Tripos Inc.
NSF National Science Foundation (Fundação Nacional de Ciência)
PDB Protein Data Bank (Banco de dados de proteínas)
PDBQT Arquivo PDB acrescido de cargas parciais (Q) e número de torsões (T)
PPGIT Programa de Pós-Graduação em Inovação Terapêutica
SDF Structure Data File (Arquivo de dados da estrutura)
SDF Site Definition File (Arquivo de definição do grid computacional local)
SDK Software Development Kit (Kit de desenvolvimento de software)
SH Shell Script (linguagem de programação)
UFPE Universidade Federal da Pernambuco

SUMÁRIO
1 INTRODUÇÃO ............................................................................................................................1
2 REVISÃO DE LITERATURA...........................................................................................................5
2.1 Modelagem Molecular ...........................................................................................................5
2.1.1 Docking Molecular...............................................................................................................5
2.1.1.1 AutoDock ..........................................................................................................................9
2.1.1.2 AutoDock Vina ................................................................................................................12
2.2 Computação Distribuída .......................................................................................................13
2.2.1 Grid Computacional ...........................................................................................................15
2.2.2 OurGrid .............................................................................................................................18
2.2.3 Aplicações de grids computacionais ..................................................................................22
2.2.3.1 BOINC .............................................................................................................................22
2.2.3.2 XSede..............................................................................................................................24
2.3 Grids computacionais aplicados em áreas diversas ..............................................................25
2.4 Docking molecular utilizando arquiteturas distribuídas .......................................................26
3 OBJETIVOS ...............................................................................................................................32
3.1 Geral .....................................................................................................................................32
3.2 Específicos ............................................................................................................................32
4 METODOLOGIA .......................................................................................................................33
4.1 GriDoMol ..............................................................................................................................33
4.2 Testes de desempenho computacional (“performance”) .....................................................36
4.2.1 Programa AutoDock ..........................................................................................................42
4.2.2 Programa AutoDock Vina ..................................................................................................43
5 RESULTADOS E DISCUSSÕES ....................................................................................................45
5.1 O programa GriDoMol ..........................................................................................................45
5.2 Resultados dos cálculos de docking......................................................................................53
5.2.1 Resultados do AutoDock ...................................................................................................55

5.2.1.1 Reprodutibilidade do tempo de execução do AutoDock ................................................63
5.2.2 Resultados do AutoDock Vina ...........................................................................................70
5.2.2.1 Reprodutibilidade do tempo de execução do AutoDock Vina ........................................77
5.2.2.2 Resultados da reordenamento da sequência de tarefas no job .....................................84
6 CONCLUSÕES ...........................................................................................................................90
7 PERSPECTIVAS .........................................................................................................................92
8 REFERÊNCIAS ...........................................................................................................................93
9 APENDICE A .............................................................................................................................98
10 APENDICE B .........................................................................................................................103

1
1 INTRODUÇÃO
O planejamento de fármacos tem à disposição as ferramentas
computacionais necessárias para a redução do tempo e diminuição dos custos
envolvidos no processo de desenvolvimento dos produtos terapêuticos ligados
à inovação.
A modelagem molecular de fármacos está associada ao estudo in silico
de estruturas e propriedades químicas de moléculas de interesse farmacêutico,
de forma a permitir, por exemplo, uma análise entre atividade biológica e
propriedades físico-químicas através de procedimentos computacionais.
Estima-se que a utilização de métodos in silico no planejamento de fármacos
pode reduzir os custos e o tempo de desenvolvimento de um novo fármaco em
até 50% (GELDENHUYS et al., 2006; MCGEE, 2005). Isto ocorre porque
muitas vezes o número de moléculas que precisam ser sintetizadas e testadas
experimentalmente passa a ser drasticamente reduzido por conta da alta
preditividade e confiabilidade dos métodos computacionais (in silico),
abreviando assim o tempo de desenvolvimento de um novo fármaco. O docking
molecular, um método da modelagem molecular, determina se há interação
energética favorável entre estas duas moléculas (ligante e alvo biológico), no
intuito de elucidar as razões moleculares responsáveis pela afinidade entre
estes ligantes (fármacos em potencial) e o sítio ativo do receptor (alvo
biológico). Desta forma, as indústrias farmacêuticas investem cada vez mais na
aquisição de computadores mais eficientes para seus centros de pesquisa
(MCGEE, 2005).

2
O procedimento de docking molecular busca pela posição e orientação
que maximiza essas interações intermoleculares. Assim, o ligante e o receptor
formam um complexo por complementaridade estrutural e por estabilização
energética. Um dos principais objetivos dos estudos na área de planejamento
in silico de fármacos é prever a intensidade e a especificidade com que
pequenas e médias moléculas, normalmente denominadas de ligantes (drogas
ou fármacos, em potencial) se liguem ao sítio ativo de um receptor biológico,
tipicamente uma bio-macromolécula (alvo farmacológico), modificando assim o
seu ciclo bioquímico/farmacológico, através de modulação da sua resposta
biológica.
Geralmente, o estudo de docking molecular ocorre na situação de se
conhecer apenas o alvo biológico e, para descobrir o ligante mais adequado
para este alvo é necessária a realização de docking molecular em larga escala,
comumente denomina-se nestes casos o termo virtual screening, entre o
receptor e um grande número de ligantes para descobrir quais deles tem maior
afinidade para interagir de maneira a formar um complexo estável com o
receptor. Ocorre que estes métodos podem apresentar, por vezes, uma alta
demanda computacional quando o número de ligantes e alvos a serem
testados é alto e quando se busca alta precisão nos resultados numéricos
obtidos.
Segundo W. Cirne, um grid computacional é uma rede de computadores
independentes em que indivíduos se conectam para usufruir maior poder
computacional para realizar tarefas de forma paralela, sem a necessidade de

3
conhecer os detalhes técnicos do grid computacional em questão (CIRNE,
2002).
A ideia de aplicar tecnologias de computação distribuída, como por
exemplo, grids computacionais para a formação de verdadeiros “laboratórios
virtuais” é atualmente é muito explorada mundo afora em projetos de diversas
áreas do conhecimento humano, inclusive em modelagem molecular. Neste
contexto, é possível encontrar iniciativas de estudos de docking molecular
acelerados por poderosas infraestruturas computacionais (BUYYA et al., 2002;
ABREU et al., 2010; ZHANG et al., 2008; JIANG et al., 2008; DOMINGUEZ;
BOELENS; BONVIN, 2003; DE VRIES; VAN DIJK; BONVIN, 2010). Desta
forma, o tempo total necessário para obtenção dos resultados de aplicações
desta natureza pode ser significativamente reduzido, uma vez que cada
execução de docking molecular será distribuído nas máquinas pertencentes a
um grid computacional, de forma abstraída para o usuário final, ou seja, este
usuário apenas submete os jobs (aplicações paralelas contendo as tarefas
individuais), enquanto o ambiente de grid computacional se encarrega de
distribuí-los e executá-los de maneira otimizada através das máquinas que
constituem o sistema integrado, passando a impressão que toda a execução
está sendo realizada na máquina do usuário.
O objetivo deste trabalho é obter uma plataforma unificada e intuitiva
para execução de docking molecular em grande escala com versões para os
sistemas operacionais mais utilizados no meio acadêmico, Linux e Windows.
Esta plataforma está apta para ser usada em conjunto com dois dos programas
“código livre” (opensource) de docking molecular mais utilizados atualmente, o

4
AutoDock (GOODSELL; MORRIS; OLSON, 1996; MORRIS et al., 2009) e o
AutoDock Vina (TROTT; OLSON, 2010).
As execuções dos cálculos de docking são realizadas em paralelo, no
ambiente de grid computacional OurGrid (ANDRADE et al., 2003). Nesta
interface, o usuário poderá lidar com todos os componentes do grid
computacional de forma prática e abstraida, e assim, manter o foco nos dados
químicos sobre moléculas de interesse, na construção de seu virtual screening.

5
2 REVISÃO DE LITERATURA
2.1 Modelagem Molecular
A modelagem molecular é o estudo in silico de estruturas e propriedades
químicas de moléculas. Como mencionado anteriormente, a utilização destes
métodos podem reduzir significativamente o tempo e os custos necessários
para o desenvolvimento de novos fármacos (GELDENHUYS et al., 2006). Um
exemplo da importância destes estudos é que a utilização desta abordagem
ajudou a encontrar inibidores do HIV (WLODAWER, 2002; DESJARLAIS et al.,
1990).
Suas aplicações vão desde a criação/edição (MDL ISISDraw, 2012;
XDrawChem, 2012) e visualização (DELANO, 2004;2009; SAYLE;
MILNERWHITE, 1995) de moléculas, até otimização de geometrias
(PURANEN; VAINIO; JOHNSON, 2010; HESS et al., 2008) e interações entre
alvos biológicos e seus potenciais ligantes, como é o caso do docking
molecular (GOODSELL; MORRIS; OLSON, 1996; MORRIS et al., 2009;
TROTT; OLSON, 2010).
2.1.1 Docking Molecular
O docking molecular é uma das técnicas da modelagem molecular que
busca obter com precisão as condições estruturais e energéticas para a
interação favorável entre um determinado alvo biológico (receptor) e uma dada
molécula (ligante). Este tipo de interação tem como objetivo formar um
complexo estável entre ambas as moléculas (LENGAUER; RAREY, 1996), o
que frequentemente leva a um efeito modulador no âmbito dos processos

6
biológicos ou farmacológicos. Os alvos biológicos mais típicos são de natureza
protéica, apesar de poder ser também de outros tipos, como ácidos nucléicos,
por exemplo. Um exemplo de interação para a formação de um complexo
(receptor + ligante) pode ser observado na Figura 2.1.
Figura 2.1 – Exemplo de um complexo estrutural (PDB: 1ULB) formado por um receptor (cor
laranja) e seu ligante co-cristalizado (modelo de esferas e ligações, em cores azul e branca). O
melhor resultado de re-docking molecular pode ser observado detalhadamente (modelo de
ligações, em cor vermelha), bem como sua alta superposição com a estrutura do ligante co-
cristalizado. (a) A visão panoramica da superfice do receptor, incluindo o ligante co-cristalizado
e a solução de docking. (b) Uma visão detalhada do sitio ativo do receptor.

7
Um dos principais objetivos dos estudos na área de planejamento in
silico de fármacos é prever a intensidade e a especificidade com que pequenas
moléculas, normalmente denominadas de ligantes (drogas ou fármacos, em
potencial) se ligam ao sítio ativo de um receptor biológico (região da
biomolécula onde ocorre as interações químicas entre o ligante e o alvo
biologico), tipicamente uma bio-macromolécula (alvo farmacológico),
modificando assim o seu ciclo bioquímico/farmacológico, através de modulação
da sua resposta biológica, tanto no homem quanto em outros animais ou
organismos patogênicos. Em termos gerais, esta modulação pode ser
tipicamente de dois tipos: i) agonista, ativando este receptor ou alvo
farmacológico, para que este desempenhe sua atividade biológica; ii)
antagonista, de forma que inibe a ação do receptor farmacológico evitando que
ele desempenhe sua atividade biológica, por exemplo reagindo com o substrato
natural que se liga a ele, uma vez que o sitio ativo está bloqueado. A decisão
sobre inibir ou ativar uma enzima que é eleita como potencial alvo
farmacológico, por exemplo, vai depender do mecanismo farmacológico e do
propósito terapêutico.
A obtenção das estruturas moleculares ocorre geralmente através da
utilização de métodos experimentais de difração de raios-X dos complexos
(receptor + ligante) cristalizados, que posteriormente são resolvidos
cristalograficamente e digitalizadas no formato PDB (PDB Database, 2012). É
possível visualizar as características e propriedades destas estruturas
registradas nos arquivos em formato PDB, através de programas projetados
especificamente para estes propósitos, tais como o AutoDock Tools (ADT,

8
2012), o Rasmol (SAYLE; MILNERWHITE, 1995), e o PyMol (DELANO,
2004;2009), dentre outros.
Durante a execução do docking molecular, tipicamente na região mais
importante do receptor, denominada sítio ativo, onde ocorre a reação com o
substrato natural, a molécula do ligante será posicionada pelo programa de
docking molecular na tentativa de ser encontrada a posição e orientação que
favoreça a ligação desta com o receptor biológico, levando em consideração
suas estruturas e a função de pontuação para determinar a estabilidade do
complexo.
Nos programas de docking molecular que utilizam uma função de
pontuação (score) baseada em campos de força (force field), que é o caso dos
programas AutoDock e AutoDock Vina, tal estabilidade da ligação ligante-
receptor é medida pela intensidade da energia de interação das forças
intermoleculares envolvidas na ligação entre os átomos do ligante e os átomos
do sítio ativo do receptor. Estas forças intermoleculares são fracas comparadas
com as forças intramoleculares (ligações covalentes) e são, em geral,
reversíveis. Quanto menor (mais negativa) for a energia de interação, maior é a
estabilidade do complexo (receptor + ligante). Como exemplo, costuma-se
observar nos resultados de cálculos, que uma interação de ligação de
hidrogênio é, tipicamente, cerca de 10 vezes mais estável do que uma
interação do tipo van der Waals. Portanto, o número de interações
intermoleculares e os tipos e intensidades das mesmas vão ter profunda
influência sobre a estabilidade de interação calculada (através do programa de
docking molecular) entre o ligante e o receptor de interesse. É importante

9
ressaltar que apenas determinadas moléculas se “encaixam” no sítio ativo do
alvo biológico, muitas vezes por razões de tamanho e volume disponíveis. Esta
estratégia de busca pela interação ligante-receptor é amplamente associada à
idéia da “chave-fechadura” (FISCHER, 1894), onde apenas a chave apropriada
está apta a se encaixar na fechadura. Um avanço do modelo “chave-
fechadura”, de Fischer, foi proposto por Koshland (KOSHLAND, 1958), que
considera um “ajuste induzido” do receptor (enzima) pela presença de uma
molécula diferente do seu substrato natural, tipicamente um modulador artificial
candidato a fármaco, por exemplo.
O docking molecular é um excelente método para avaliar os possíveis
ligantes de uma proteína de interesse, uma vez que tem a vantagem de utilizar
a abordagem “in silico” que dispensa a necessidade de reproduzir
exaustivamente em laboratório cada possibilidade de ligação. Assim, apenas
as moléculas que demonstrem mais condições de uma ligação estável serão
sintetizadas em laboratório, a posteriori, aumentando a capacidade de teste e
economizando tempo e dinheiro (GELDENHUYS et al., 2006).
2.1.1.1 AutoDock
O programa AutoDock é uma ferramenta open-source para realização de
docking molecular, que se baseia em cálculos computacionais para obter a
energia de interação mínima para o complexo ligante-receptor, explorando os
principais graus de liberdade das moléculas de interesse (GOODSELL;
MORRIS; OLSON, 1996; MORRIS et al., 2009).

10
Antes da execução deste programa, é necessário converter o formato dos
arquivos das moléculas para o formato PDBQT, que é um formato estendido do
PDB onde estão inclusas, além das informações estruturais da molécula
contidas no formato PDB, as cargas elétricas de cada átomo que constitui a
molécula (Q) e os tipos e números de torções (T). A geração do arquivo
PDBQT é obtida por meio da utilização do programa utilitário AutoDock Tools
(ADT, 2012), que faz parte do pacote de ferramentas computacionais
compatíveis com o programa Autodock, permitindo que os usuários realizem
todo o processamento dos arquivos e ajustes para a execução dos cálculos de
docking molecular.
Para a realização do cálculo de docking no programa AutoDock, é
necessário pré-calcular os mapas de energia de interação para os tipos de
átomos que compõe os ligantes de interesse. Estes mapas são construídos a
partir da simulação de sondas atômicas que varrem tridimensionalmente a
região definida como a mais importante para a busca das soluções de docking
(poses), particularmente no sítio ativo do alvo farmacológico, quando esta
informação é conhecida. O programa AutoGrid (que faz parte do pacote de
programas do AutoDock) é utilizado para este mapeamento energético. Por
exemplo, se o ligante possui um átomo de carbono do tipo aromático e um
oxigênio aceitador de ligação de hidrogênio, estes átomos serão usados na
varredura tridimensional na região do sitio ativo e as energias de interação de
cada um destes átomos com os átomos do sitio ativo do receptor serão
mapeadas (A.map e OA.map, respectivamente).

11
O objetivo deste pré-cálculo com o módulo AutoGrid é tornar o cálculo
de docking mais rápido e eficiente como um todo, uma vez que não haverá a
necessidade de recalcular as energias de interação dos átomos envolvidos
durante o processo de docking, realizado efetivamente pelo módulo AutoDock.
O mapeamento eletrônico para o cálculo do potencial eletrostático também é
realizado através do módulo AutoGrid. Está etapa inicial é obrigatória, uma vez
que o módulo AutoDock necessita destes mapas para a sua execução, na
próxima etapa do cálculo.
Antes de realizar a execução, tanto do AutoGrid quanto do AutoDock, é
necessário gerar os arquivos de configuração, que comandam detalhadamente
a realização dos cálculos executados por cada um destes programas. Estes
arquivos de configuração são denominados, grid parameter file (gpf) e docking
parameter file (dpf), para os programas AutoGrid e AutoDock, respectivamente.
A criação destes arquivos de configuração é realizada através da utilização da
interface gráfica AutoDock Tools (ADT, 2012). Estes arquivos de configuração
contêm informações importantes sobre como serão efetuadas as buscas pela
melhor solução estrutural e energética de docking. Dependendo dos critérios
usados para a edição destes arquivos, é possível tornar os cálculos de docking
mais precisos, porém com alta demanda computacional para cada cálculo
individual de docking molecular, chegando a impactar o tempo necessário para
o término do processo em até algumas ordens de magnitude.
De posse das moléculas em seu formato PDBQT e dos mapas atômicos
gerados pelo programa AutoGrid, é possível realizar o cálculo de docking
molecular usando-se o programa AutoDock. Utilizando uma combinação de

12
algoritmos genéticos, principalmente o LGA (Lamarckian Genetic Algorithm), o
programa AutoDock busca pela melhor solução de docking entre receptor e
ligante.
2.1.1.2 AutoDock Vina
O programa AutoDock Vina (TROTT; OLSON, 2010) é uma versão
adaptada do programa AutoDock (GOODSELL; MORRIS; OLSON, 1996;
MORRIS et al., 2009) com suporte a execução multicore, com o intuito de
usufruir da disponibilidade de todos os núcleos de processamento presentes
nos computadores atuais. Além disto, nesta versão modificada do AutoDock, o
processo de realização do docking molecular se tornou mais simples,
automático e intuitivo em comparação ao AutoDock, uma vez que requer
apenas as estruturas das moléculas que serão usadas no docking molecular,
dispensando a etapa do cálculo com o módulo AutoGrid.
O docking molecular com o programa AutoDock Vina pode ocorrer em
diversos núcleos de processamento existentes na máquina para paralelizar a
carga ou demanda computacional dos cálculos envolvidos no procedimento, a
fim de acelerar o processo como um todo, com resultados bastante
satisfatórios de desempenho. A estratégia de paralelismo utilizada pelo
AutoDock Vina consiste em dividir a superfície de busca de seus algoritmos
genéticos em diversas subáreas de cálculos para distribuí-las nos diversos
núcleos de processamento da máquina. Assim, cada núcleo de processamento
realiza a evolução das populações em uma determinada região específica do
domínio de buscas e no final, os complexos receptor-ligante mais estáveis

13
encontrados durante a evolução das populações, em cada subárea, são
reunidos e ordenados de forma crescente em relação aos valores de energias
de interação calculados. Esta estratégia torna o programa AutoDock Vina
consideravelmente mais rápido do que o programa AutoDock, sendo até 62
vezes mais rápido (TROTT; OLSON, 2010).
2.2 Computação Distribuída
A computação distribuída consiste na utilização de técnicas para unir a
capacidade de processamento de vários computadores para a execução de
uma tarefa complexa que esteja relacionada a uma elevada demanda
computacional.
Existem diversos tipos de arquiteturas distribuídas, sendo que os mais
comumente usados são o cluster e o grid computacional. Embora sejam
similares no comportamento, ambas possuem suas vantagens e desvantagens
(ver comparação na tabela 1).
Tabela 1 – Comparativo entre alguns pontos chaves entre cluster e grid computacional
(SADASHIV; KUMAR, 2011).
Cluster Grid Computacional
Máquinas homogêneas Máquinas heterogêneas
Dentro dos limites institucionais Podem estar distribuídos geograficamente
Controle centralizado, todo processamento
das máquinas disponíveis são dedicados à
máquina central (front-end), tornando-as
uma única máquina robusta
Cada máquina possui autonomia, podendo
ser utilizada para outros propósitos quando
não estiver integrando o grid.

14
Um dos ambientes de grid computacional mais conhecido e utilizado no
mundo é o BOINC (BOINC, 2012), desenvolvido pela Universidade de
Berkeley. A média diária de poder de processamento neste ambiente de grid
computacional é de aproximadamente 10 petaFLOPS, ou seja, 10 quadrilhões
de operações de ponto flutuante por segundo. Este poder de processamento
supera o obtido pelo supercomputador (cluster) mais rápido do mundo, em
junho de 2011, o K Computer. Este supercomputador, que utiliza a arquitetura
SPARC64 da Fujitsu, na época possuía 548.352 núcleos de processamento e
seu poder de processamento era de aproximadamente 8.2 petaFLOPS
(TOP500 Supercomputers, 2012). Atualmente, ele se encontra na terceira
posição possuindo 705.024 núcleos de processamento e tem o poder
computacional de 10.6 petaFLOPS, muito próximo do obtido no ambiente de
grid computacional BOINC. Em uma análise mais atualizada, o
supercomputador mais rápido do mundo, nos dias de hoje, é o Titan (TOP500
Supercomputers, 2012). Este supercomputador, que utiliza a arquitetura Cray
XKY da Cray, possui 560.640 núcleos de processamento e seu poder
computacional é de aproximadamente 17,6 petaFLOPS.
Como, geralmente, a infraestrutura computacional de um ambiente de
grid computacional é proporcionada por doações voluntárias dos ciclos de
processamento ociosos das máquinas dos usuários, em suas casas ou
ambientes de trabalho, os custos de manutenção e consumo energético destas
máquinas são arcados por estes próprios usuários, tornando o acesso a este
poder computacional muito mais barato do que manter um supercomputador.
Vale ressaltar que, na maioria dos casos, grids computacionais, são acessíveis
a qualquer pessoa ou usuário que tenha interesse em participar deste tipo de

15
ambiente distribuído, ao contrário de muitos supercomputadores que são de
uso restrito de empresas ou outras instituições.
Outro meio de utilização de arquiteturas distribuídas é em nível de
núcleos de processamento. Nos processadores atuais, podemos observar que
muitos deles possuem mais de um núcleo de processamento, como, por
exemplo, em processadores dual-core ou quad-core, onde é possível distribuir
uma tarefa entre estes núcleos de processamento do processador, comumente
denominados CPU. Um bom exemplo, que utiliza esta estratégia para
aumentar o desempenho computacional da aplicação, é o programa AutoDock
Vina (TROTT; OLSON, 2010). O mesmo ocorre com as placas gráficas atuais,
onde através de uma linguagem de programação especializada (CUDA, 2012),
pode-se usar estes núcleos de processamento gráfico, comumente
denominados GPU, para acelerar a execução do programa de maneira análoga
ao que ocorre a nível de CPUs. Esta abordagem vem sendo amplamente
usada em vários projetos na área de modelagem molecular como é o caso do
programa de busca “as cegas” pelo sitio ativo (SUKHWANI; HERBORDT,
2010) e até mesmo no campo do docking molecular (ROH et al., 2009).
Devido a sua relevância neste presente trabalho, iremos detalhar os
conceitos sobre grid computacional e sua aplicabilidade no meio científico.
2.2.1 Grid Computacional
Ambientes de grids computacionais são tipicamente formados por um
conjunto de computadores, autônomos e geralmente heterogêneos, ou seja,
máquinas com configurações de hardware e software diferentes entre si,
interconectados em larga escala, e que podem, até mesmo, estar distribuídos

16
geograficamente para atender uma demanda computacional elevada (SMITH,
2004). O papel principal deste conjunto de computadores autônomos é realizar
tarefas complexas que são inviáveis de serem executadas em apenas uma
única máquina. Além da sua capacidade de processamento, todos os recursos
das máquinas envolvidas podem ser compartilhados entre si, tais como sua
capacidade de armazenamento, sensores e outros periféricos ligados a elas.
Desta forma, é possível obter um poder computacional muito grande, de forma
abstraida para o usuário do grid computacional. Uma analogia pode ser feita
com as redes de telefonia ou de eletricidade. Em ambas, o usuário não precisa
conhecer exatamente onde está localizado fisicamente cada componente das
redes mencionadas para utilizar seus recursos (CIRNE, 2002). Desta forma,
assim como é conectado um equipamento elétrico na tomada para obter a
energia elétrica, basta conectar um computador a um grid computacional, para
ter acesso aos recursos dos computadores distribuídos ao longo de toda a
estrutura de computação distribuída.
Empresas e instituições que fazem parte de um ambiente de grid
computacional doam ciclos de processamento de seus computadores em
período ocioso para, quando precisarem, poderem ter acesso a mais recursos
dos outros membros do grid computacional. As principais características de um
grid computacional é abstrair os detalhes técnicos ao utilizar os recursos que
podem estar, inclusive, distribuídos geograficamente (Figura 2.2) e a ausência
de um controle central para todo o ambiente de grid computacional. Para
atender as demandas computacionais, é esperado um bom nível de Qualidade
de Serviço (Quality of Service, QoS) (FOSTER, 2002), tais como
disponibilidade, segurança e tempo de resposta.

17
Figura 2.2 – Heterogeneidade e distribuição geográfica de um grid computacional (UK Total,
2012). Extraido de http://www.uk.total.com/activities/innovative_computing.asp
O conceito proposto para tentar definir os principais aspectos da
computação em grid que a diferencia das demais estruturas de computação
distribuída são (CIRNE, 2002):
A heterogeneidade dos componentes do grid computacional, uma vez que
consegue realizar a interação entre computadores que podem utilizar
arquiteturas de hardwares e sistemas operacionais diferentes.
A dispersão geográfica do grid computacional, levando em consideração
que os recursos computacionais de um grid podem estar distribuídos em
todo o mundo, não se limitando a barreira física institucional.
O compartilhamento dos recursos do grid computacional: todo poder
computacional do grid está disponível para ser utilizado em diversos
propósitos.

18
Múltiplos domínios administrativos em um ambiente de grid computacional,
uma vez que algumas instituições podem doar e receber recursos
computacionais para outras instituições.
O controle distribuído, caracterizando a ausência de um mecanismo de
controle único e centralizado para todo o grid computacional.
Um exemplo que expressa à idéia de um grid computacional simples é a
própria internet, onde são utilizados recursos de servidores externos para obter
dados ou processamento (CIRNE, 2002). Não é preciso saber o caminho feito
a cada roteador e nem a localização física de cada servidor para usufruir das
funcionalidades destes vários servidores na internet.
2.2.2 OurGrid
O projeto OurGrid é uma iniciativa nacional de implementação de um
ambiente de grid computacional, desenvolvido pela Universidade Federal de
Campina Grande (UFCG) desde 2003 (ANDRADE et al., 2003). O OurGrid
partilha da filosofia “opensource” e é escrito na linguagem de programação
Java, o que permite que todos os seus componentes possuam versões para
todos os sistemas operacionais que suportem a plataforma Java. Uma lista
contendo alguns projetos que utilizam a plataforma OurGrid de ambiente grid
computacional pode ser encontrada no site oficial do projeto (OurGrid, 2012).
O OurGrid é voltado para a execução de tarefas que utilizam o modelo
BoT (bag-of-tasks), ou seja, os processos em paralelo que não precisam se
comunicar entre si, por apresentarem independência lógica e temporal. Embora
as tarefas BoT sejam em sua grande maioria simples, elas são muito utilizadas

19
em diversos cenários científicos (CIRNE et al., 2003), o que aumenta a
aplicabilidade deste sistema para aplicações científicas.
A infraestrutura de rede utilizada é a peer-to-peer (p2p), que é uma
infraestrutura descentralizada e heterogênea de compartilhamento de recursos,
onde não há a necessidade de um servidor central para controlar os
integrantes da rede (SCHOLLMEIER, 2002). Sendo assim, todos os nós
integrantes da rede podem compartilhar seus recursos, mesmo possuindo
diferentes arquiteturas de hardware e software. Para garantir uma distribuição
justa dos recursos, o OurGrid utiliza um mecanismo chamado Network of
Favours, de modo que a empresa ou instituição que contribuir cedendo mais
recursos terá direito a mais recursos alocados quando necessitar de mais
poder computacional. Desta forma o compartilhamento de recursos
computacionais é amplamente incentivando.
O método mais simples para obter acesso ao poder computacional
disponível no ambiente de grid computacional do projeto OurGrid é se
registrando no OurGrid Portal (OurGrid Portal, 2012). Porém, desta forma, não
é possível contribuir com recursos próprios para o ambiente de grid
computacional e, consequentemente, a prioridade para a execução das tarefas
(tasks) contidas no job do usuário será baixa, por causa do mecanismo
Network of Favours. Para aumentar a prioridade de execução das tarefas
(tasks) e obter acesso a maior quantidade de máquinas, o usuário deve instalar
seu próprio grid computacional contendo os componentes da plataforma
OurGrid. Neste caso, pode-se optar entre integrar o grid computacional do
projeto OurGrid, através da opção join the community nas configurações do

20
componente Peer, ou se deseja apenas criar um ambiente de grid
computacional pessoal ou institucional. Os três componentes responsáveis
pela gestão e comunicação do ambiente OurGrid são:
O componente Broker é o responsável por escalonar as tarefas (tasks) ao
longo dos computadores (Workers) disponíveis no ambiente de grid
computacional. As principais interações entre o usuário e o ambiente de grid
computacional ocorrem através deste componente. Este componente
necessita ter acesso a, pelo menos, um Peer para iniciar suas atividades.
O componente Peer é o responsável por gerenciar os computadores
pertencentes ao grid computacional de certa empresa ou instituição. Este
componente identifica e disponibiliza as máquinas ociosas (Workers) para a
execução remota de uma dada tarefa. Desta forma ele é responsável por
autorizar e realizar a comunicação entre Brokers e Workers.
O componente Worker é responsável pela execução das tarefas recebidas
remotamente. Para isso, este componente recebe os arquivos de entrada e
o programa que será utilizado para realizar o processamento e
posteriormente retorna o resultado deste processamento de volta para a
máquina do usuário que requisitou a execução. Este componente está
presente em cada computador integrante do grid computacional que esteja
apto para ceder seus recursos computacionais.
Para realizar a comunicação entre os componentes do ambiente de grid
computacional OurGrid, é necessário utilizar o protocolo XMPP. Este protocolo

21
detecta a presença destes componentes e possibilita a troca de mensagens
entre eles.
O conjunto de tarefas (tasks) do OurGrid é denominado job. Para
realizar a execução destes jobs, o OurGrid utiliza uma estrutura de arquivo
chamada JDF (Job Description File), na qual é definido os arquivos de entrada
(input) e os de saída (output) bem como a execução, e os parâmetros que
serão utilizados.
Basicamente, cada task é subdividida em três partes (init, remote e
final), as quais indicam a máquina remota como proceder de acordo com a fase
em que a tarefa (task) se encontra. A fase init, que corresponde ao início,
consiste em transferir os arquivos necessários para a execução do job, bem
como o programa utilizado e seus arquivos de entrada (input). A fase remote,
consiste na realização da execução em si. Nela, constará o comando que será
realizado na máquina remota. Há também uma fase chamada final, que
consiste no retorno dos resultados (output) gerados pela execução da tarefa.
Antes de definir as tarefas, é necessário iniciar as configurações padrão, ou
seja, comum a todas as atividades (tasks). No inicio do arquivo Job Description
File, Após a tag “job”, é possível especificar o label (nome que será exibido
durante sua execução) e os requisitos mínimos necessários que o computador
remoto precisa para realizar a execução do job, como por exemplo, qual
sistema operacional, quantidade de memória RAM, espaço em disco, dentre
outros. Também há a possibilidade de definir etapas padrões para as tarefas,
em qualquer uma das fases init, remote e final, e assim as tarefas que não

22
possuam registro de alguma destas fases, usará o que foi definido como
padrão.
Os comandos usados para manipular os arquivos entre a máquina do
usuário e o ambiente de grid computacional são o put, store e o get. Os
comandos put e store armazenam, no ambiente de grid computacional, os
arquivos necessários para a execução das tarefas (tasks). A diferença entre
estes dois comandos está no tempo em que o arquivo permanece na máquina
remota (Worker). O comando store armazena permanentemente o arquivo na
máquina remota (Worker), ideal na situação onde o arquivo necessita ser
utilizado mais de uma vez, enquanto o comando put armazena o arquivo
somente até o termino da execução, ideal para arquivos que serão utilizados
poucas vezes. O comando get retorna o arquivo resultante da execução remota
de volta para a máquina do usuário.
2.2.3 Aplicações de grids computacionais
As motivações para o uso de um ambiente de grid computacional podem
variar entre os modelos voltados a propósitos sociais, como analises climáticas
e previsão de terremotos, até modelos comerciais e modelos moleculares.
2.2.3.1 BOINC
A plataforma opensource para computação voluntaria BOINC (BOINC,
2012) é uma ferramenta que forma um grid computacional através de
computadores comuns, disponibilizados voluntariamente pelos usuários que

23
instalam seu software, voltado para pesquisa cientifica. A escolha dos projetos
que serão beneficiados pelos ciclos de processamento doados fica ao encargo
do próprio usuário, através da extensa lista de projetos suportados pelo
BOINC. Além da possibilidade de fornecer o poder computacional voluntário,
há também a opção de criar novos projetos dentro da plataforma BOINC e,
desta forma, se beneficiar do poder computacional de outros usuários.
O processamento dos dados pode ser feito através dos convencionais
núcleos de processamento do processador (CPU) como também utilizar os
núcleos de processamento gráfico (GPU), em opções mais recentes.
Para incentivar a colaboração, é disponibilizado um website com uma
tabela de pontuação e classificação dos usuários, indicando quais colaboraram
mais ativamente no desempenho do grid computacional e seus respectivos
projetos. O serviço conta com, atualmente, mais de 250 mil voluntários ativos e
mais de 350 mil computadores.
Como pode ser observado na lista de projetos ativos (Choosing BOINC
projects, 2012), os projetos possuem suporte a diversos sistemas operacionais,
indo desde sistemas desktops convencionais como Windows, Mac e Linux, até
sistemas um pouco mais recentes, como, por exemplo, o Android.
O World Community Grid (World Community Grid, 2012) é um ambiente
de grid computacional que emprega a plataforma BOINC para utilizar os ciclos
de processamento doados por voluntários. Os projetos disponibilizados neste
website são focados para benefício humanitário. Alguns destes projetos são
voltados para a área da saúde e executam o programa AutoDock Vina na
máquina do voluntário, na tentativa de encontrar um potencial fármaco para

24
algumas das doenças estudadas. Entre os projetos ativos que utilizam esta
abordagem estão Say No to Schistosoma (Say No to Schistosoma, 2012), GO
Fight Against Malaria (GO Fight Against Malaria, 2012) e Drug Search for
Leishmaniasis (Drug Search for Leishmaniasis, 2012).
2.2.3.2 XSede
Sucessor do agora descontinuado TeraGrid, o Xsede (Xsede, 2012) é
um projeto que utiliza, em larga escala, computadores e bancos de dados para
realização de pesquisas cientificas. É coordenado pelo NSF, National Science
Foundation (NSF, 2012), e é uma parceria entre diversas instituições
acadêmicas dos Estados Unidos.
De forma geral, para utilizar seus recursos, o usuário precisa se
cadastrar e enviar uma proposta para a utilização de um determinado recurso.
Estas requisições podem ser:
Iniciante (startup): Alocação recomendada para os novos usuários. É um
nível limitado, e seu uso é ideal para demonstrações em sala de aula e para
conhecer sua infraestrutura. Pode ser requisitado por funcionários e
pesquisadores pós-doutorados em universidades dos Estados Unidos como
também por professores do ensino fundamental e médio americanos. Este
nível tem a limitação de um ano de duração, podendo ser renovado após o
termino, e 200 mil CPU-horas de processamento.
Educação (education): Comumente utilizado para aulas acadêmicas e
treinamentos, este nível é apropriado quando se tem datas fixadas para inicio e

25
termino da atividade. Possui os mesmos limites que o nível iniciante,
diferenciando do mesmo apenas pela natureza e propósito do estudo.
Pesquisa (research): Para usuários que necessitam de mais recursos do
que os que estão disponíveis nos níveis anteriores. A alocação dos recursos é
feita sob medida e é concedido de acordo com a meritocracia. A requisição no
nível de pesquisa só é aberta uma vez a cada trimestre.
Independentemente do tipo de alocação almejado pelo pesquisador, não
há custo algum para usufruir o poder computacional disponível neste sistema,
tornando sua infraestrutura bastante acessível.
2.3 Grids computacionais aplicados em áreas diversas
O projeto ImmunoGrid (PAPPALARDO et al., 2009) utiliza o ambiente
distribuído de um grid computacional colaborativo para a descoberta,
formulação e otimização de vacinas. Seu funcionamento consiste em
simulações computacionais, distribuídos em larga escala em um ambiente de
grid computacional, das interações entre antígenos e o sistema imunológico.
Estas interações são calculadas através do uso de modelos matemáticos de
equação diferencial e autómato celular para modelar as interações entre as
moléculas e as células responsáveis pela resposta do sistema imunológico. O
ImmunoGrid possui um portal web voltado a pesquisa e aprendizado. Porém, o
acesso a estes recursos é restrito a projetos selecionados pela equipe do
ImmunoGrid. Durante os testes de desempenho neste ambiente de grid
computacional, foram simuladas 1600 vacinas em 100 camundongos,

26
representando os 24 meses de vida de cada camundongo, em apenas 26
horas.
No estudo realizado por Christopher Woods (WOODS et al., 2005), um
dos projetos realizados envolve o uso de grids computacionais para realizar
estudos de dinâmica molecular. Neste projeto, montou-se um ambiente de grid
computacional com 450 máquinas heterogêneas e não dedicadas, espalhadas
por toda a universidade de Southampton, e integradas através do ambiente
Condor (HTCondor, 2013). Realizou-se múltiplos cálculos de dinâmica
molecular com a proteina NtrC (proteína C reguladora de nitrogênio) em
diversas condições, como por exemplo, de temperatura. Isto resultou no total
de 64 execuções sendo realizadas em paralelo no ambiente de grid
computacional. Foi utilizado um cluster adicional neste tipo de sistema, que
melhorou a eficiencia do ambiente de grid computacional de 63 para 78% do
tempo ativo.
2.4 Docking molecular utilizando arquiteturas distribuídas
O Dovis (ZHANG et al., 2008) é um sistema que utiliza o ambiente
distribuído de um cluster para realizar virtual screening em larga escala através
de uma interface gráfica. O programa de docking molecular utilizado neste
estudo foi o AutoDock (GOODSELL; MORRIS; OLSON, 1996). Este sistema
HPC, high-performance computing, é capaz de realizar, em cada unidade de
processamento (CPU), entre 500 a 1000 cálculos de docking molecular por dia.
Seu funcionamento pode ser descrito em três etapas, sendo elas:

27
Pré-docking: Nesta etapa, ocorre a conversão das moléculas
selecionadas, do formato PDB (ou MOL2), para o formato PDBQT, o
qual é o formato padrão para a execução do cálculo de docking
molecular através do AutoDock. Após esta conversão de arquivos, as
moléculas são dividas em N subgrupos, sendo N o número de
processadores disponíveis no cluster e cada subgrupo tendo
aproximadamente o mesmo número de moléculas.
Docking molecular em paralelo: Antes do procedimento de docking
molecular, é efetuado, para cada receptor, o cálculo dos mapas da
grade de energia (utilizando o programa AutoGrid, explicado na seção
do 2.1.1.1 AutoDock) de cada tipo de átomo presente nos ligantes que
irão interagir com o receptor em questão. Após estes cálculos, é
efetuado o docking molecular e os resultados, baseados nas energias de
interação calculadas, são comparados entre os resultados dos outros
processadores, para assim, ser possível determinar os melhores
resultados globais.
Pós-docking: Os melhores resultados coletados na etapa anterior são
comprimidos e gravados em um diretório do sistema, com os resultados
finais.
Em sua versão 2.0 (JIANG et al., 2008), com o intuito de aumentar a
performance, o Dovis teve seu algoritmo de escalonamento modificado para
dinâmico, permitindo o balanceamento do cluster em tempo real. Além disso, o
código fonte do programa AutoDock também foi modificado para reduzir os
acessos ao disco rígido na escrita dos arquivos de resultados durante o
procedimento de docking molecular com este programa, uma vez que o cluster

28
em questão utilizava um disco rígido comum a todas as máquinas, e o excesso
de escrita simultânea neste disco sobrecarregava-o, tornando-o mais lento, e
consequentemente os cálculos de docking molecular demoravam mais.
Com o intuito de classificar as soluções de docking molecular mais
favoráveis dentre as encontradas nos cálculos, os programas de docking
molecular utilizam uma função de pontuação, comumente conhecida como
score, e que, geralmente, é única para cada programa. O Dovis 2.0 permite
que o usuário utilize uma função de pontuação diferente da empregada no
programa de docking molecular selecionado, procedimento este conhecido pelo
nome de re-score. Com isso, pode-se gerar uma nova classificação para os
resultados obtidos. Além disto, há a opção de converter os resultados finais
para o formato SDF, formato amplamente utilizado na modelagem molecular de
fármacos (JIANG et al., 2008).
Os testes de validação do desempenho do Dovis 1.0 com o banco de
dados ZINC, utilizando gradualmente até 128 núcleos de processamento
(CPUs), demonstrou uma progressão quase linear no ganho de desempenho
obtido. O cruzamento do banco de dados ZINC com a cadeia A da proteína
Ricina, utilizando 256 CPUs, conseguiu realizar o docking de aproximadamente
700 ligantes por CPU, ao dia (ZHANG et al., 2008).
O sistema Haddock (DOMINGUEZ; BOELENS; BONVIN, 2003; DE
VRIES; VAN DIJK; BONVIN, 2010) é um exemplo de servidor web para
docking molecular com o auxilio de dados experimentais (knowledge based)
das moléculas. Seu método de docking molecular consiste em minimização de
energia do corpo rígido, com refinamento semi-flexível dos ângulos de torção e

29
um refinamento final com solvente explicito. Possui interfaces voltadas para
cada nível de usuário:
Fácil: Requer apenas as estruturas e a lista dos resíduos do sitio ativo
que interagem com o ligante. Para todos os outros parâmetros utilizados
no procedimento de docking, é definido o valor padrão ou determinado
automaticamente.
Especialista: Permite a personalização de alguns parâmetros, tais como
a definição dos segmentos flexíveis, distâncias entre os átomos, estado
de protonação, dentre outros.
Guru: Nesta interface, além de permitir as mesmas alterações da
interface especialista, também dá acesso a configuração de constantes,
tais como, constantes de força, temperatura e pesos dos termos da
função de pontuação (score).
Todo o processamento é feito no cluster dedicado a este projeto e conta
também com acesso ao ambiente de grid computacional europeu e-NMR (e-
NMR, 2012), caso necessite de mais poder computacional.
Outro exemplo de docking molecular em sistemas distribuídos é o
sistema Mola, desenvolvido para execução de virtual screening em um
pequeno cluster heterogêneo com máquinas não dedicadas. Sua arquitetura
utiliza uma versão personalizada LIVE-CD, não necessitando instalação, do
sistema operacional Linux, o que fornece os recursos para integrar máquinas
com sistemas operacionais e hardware heterogêneos, utilizando os protocolos
LAM-MPI (LAM-MPI, 2012), Local Area Multicomputer MPI, e o MPICH
(MPICH, 2012), MPI CHameleon, ambos baseados no MPI (Message Parsing
Interface), um padrão para comunicação de dados em sistemas distribuídos.

30
Por se tratar de um LIVE-CD, não há qualquer modificação no sistema
operacional original de cada uma das máquinas utilizadas no cluster, e assim,
ao termino da execução, as máquinas voltam ao seu estado original. É
necessário que cada uma das máquinas que integram o cluster possua a
funcionalidade PXE, Preeboot eXecution Everionment, o qual permite a
inicialização do sistema a partir do comando de outra máquina ligada na
mesma rede.
Os programas de docking molecular disponíveis neste sistema são o
AutoDock (GOODSELL; MORRIS; OLSON, 1996) e o AutoDock Vina (TROTT;
OLSON, 2010), ambos desenvolvidos pelo The Scripps Research Institute
(Scripps Research Institute, 2012).
A preparação das moléculas deve ser realizada através do AutoDock
Tools (ADT, 2012), incluso no sistema operacional. No caso do AutoDock, é
necessário preparar os mapas atômicos, etapa realizada através do módulo
AutoGrid. Após a preparação inicial é possível configurar os ajustes (setups) de
docking específicos de cada programa, tais como os parâmetros do algoritmo
genético do programa AutoDock e a variável exhaustiveness, responsável pelo
número de execuções da etapa de refinamento parcial dos cálculos individuais,
no caso do programa AutoDock Vina.
Durante a execução dos cálculos de docking molecular, o sistema Mola
envia, para cada máquina, uma instância de execução do programa AutoDock
para cada CPU. No caso do AutoDock Vina, apenas uma instância é enviada
por máquina, uma vez que este programa é otimizado para utilizar todos os
núcleos de processamento disponíveis em cada máquina. Novas tarefas só são

31
atribuídas apenas quando as execuções nas máquinas finalizam, e assim,
evitando sobrecarregar máquinas mais lentas e mantendo o cluster estável.
Ao utilizar 5 máquinas dual-core, totalizando 10 CPUs, este sistema foi
capaz de realizar o docking de 237 ligantes por dia para o programa AutoDock
e 575 ligantes por dia para o programa AutoDock Vina, em cada CPU.
Portanto, o trabalho de pesquisa apresentado tem o objetivo de propor o
uso do ambiente distribuído através de um grid computacional para acelerar os
cálculos de docking molecular, por meio do desenvolvimento do programa
GriDoMol, programa este capaz de criar e submeter tarefas de docking
molecular utilizando uma interface amigável e intuitiva para o usuário final.

32
3 OBJETIVOS
3.1 Geral
Desenvolvimento e implementação de software para aplicação de grids
computacionais em modelagem para inovação terapêutica.
3.2 Específicos
Desenvolver o programa GriDoMol para gerenciar os cálculos de
docking molecular utilizados em inovação terapêutica.
Contemplar a integração dos programas de docking molecular AutoDock
e AutoDock Vina, amplamente usados no meio acadêmico.
Desenvolver versões para outros sistemas operacionais, como o Linux.
Desenvolver uma interface gráfica de usuário (GUI) amigável e intuitiva,
com o intuito de aumentar a interatividade com o usuário.
Validar o ganho de desempenho obtido ao empregar a plataforma de
grid computacional através da análise de desempenho (“benchmark”).
Tornar o GriDoMol uma plataforma unificada e intuitiva para a execução
prática de cálculos de docking molecular de forma distribuída, em um
ambiente de grid computacional, buscando alto desempenho
computacional.

33
4 METODOLOGIA
4.1 GriDoMol
O programa desenvolvido neste estudo, o GriDoMol, foi desenvolvido na
linguagem de programação C++ utilizando a biblioteca de desenvolvimento QT
(QT, 2012), amplamente utilizada no mundo acadêmico e comercial pelo seu
alto grau de desempenho e portabilidade. O QT é uma biblioteca
multiplataforma e, uma vez codificada a aplicação, ela pode ser recompilada
facilmente nas diversas plataformas que possuem a distribuição do QT. Assim,
foi possível desenvolver versões do GriDoMol para Linux e Windows com
poucas modificações no código fonte.
A interface de desenvolvimento (IDE) utilizada foi o QT Creator, incluso
no kit de desenvolvimento (sdk) do QT (QT, 2012). A escolha desta IDE se
deve ao fato de que a mesma possui versões multiplataformas, facilitando o
desenvolvimento de código fonte nestes sistemas operacionais sem a
necessidade de importar ou modificar os arquivos do projeto em si para se
adequarem a outra IDE.
O compilador utilizado no sistema operacional Windows foi o Mingw
(Mingw, 2013), também disponível no kit de desenvolvimento do QT. No
sistema operacional Linux, utilizou-se o compilador GNU C Compiler (GCC),
que é um compilador para as linguagens de programação C e C++ incluido na
maioria das distribuições Linux existentes no mercado.
Embora boa parte do código utilize as funções próprias da biblioteca de
desenvolvimento QT, houve algumas partes do código fonte que foram
comandos especificos para o sistema operacional. Nestes casos, utilizou-se os

34
recursos de definição “#ifdef Q_OS_WIN” (para Windows) e “#ifdef
Q_OS_LINUX” (para o Linux), para indicar ao compilador qual parte do código
deve ser compilado para determinada plataforma alvo e, assim, tornar o
GriDoMol compilável para estas duas plataformas utilizando um único código
fonte.
Para permitir o acesso aos componentes do OurGrid via linha de
comando no sistema operacional Windows, houve a necessidade de editar o
arquivo de lote (.bat) de cada componente do OurGrid. Houve uma modificação
na parte “(...) org.ourgrid.broker.ui.async.Main” para “(...)
org.ourgrid.broker.ui.sync.Main %1 %2”, no componente Broker, e de “(...)
org.ourgrid.peer.ui.async.Main start (...)” para “(...) org.ourgrid.peer.ui.sync
.Main %1 %2 (...)”, no componente Peer. O comando “start /b javaw” foi
substituído por apenas “java” no inicio de cada um destes arquivo de lote.
Estas modificações foram necessárias para tornar o OurGrid executável em
linha de comando.
Para realizar a comunicação entre os componentes do ambiente de grid
computacional denominado OurGrid, é necessário utilizar o protocolo XMPP.
Este protocolo detecta a presença dos componentes do OurGrid nas máquinas
e possibilita a troca de mensagens entre eles. O servidor XMPP utilizado neste
trabalho foi o OpenFire 3.7.0 (OpenFire, 2012), por se tratar do servidor
certificado e recomendado, durante o inicio deste estudo, pela equipe do
OurGrid. Atualmente, o servidor XMPP recomendado para as versões mais
recentes do OurGrid é o ejabberd (ejabberd, 2012).

35
A plataforma de grid computacional escolhida para formar o ambiente
distribuído necessário na execução das aplicações paralelas neste projeto foi o
OurGrid 4.2.3 (ANDRADE et al., 2003), por ser adequado para aplicações do
tipo BoT (Bag-of-Tasks), ou seja, aquelas onde as tarefas têm independência
temporal e lógica entre si, além de ser utilizado por outras iniciativas de
computação distribuída e de possuir uma ampla gama de aplicabilidade
(MATTOS et al., 2008; ARAUJO et al., 2005).
A escolha do programa AutoDock (GOODSELL; MORRIS; OLSON,
1996; MORRIS et al., 2009) como um dos programas para a realização de
docking molecular se deve ao fato dele ser amplamente empregado no meio
acadêmico, sendo o programa de docking molecular mais citado até janeiro de
2011. Seu artigo principal foi citado mais de 2700 vezes (AutoDock, 2012). O
programa AutoDock possui uma boa correlação entre os resultados previstos
no docking e os experimentais.
O programa AutoDock Vina (TROTT; OLSON, 2010) também foi
escolhido por ser um programa ainda mais prático e rápido do que o AutoDock.
Neste caso (AutoDock Vina), não há a necessidade de que o usuário realize o
cálculo das energias de interação no modulo AutoGrid, tornando o cálculo de
docking molecular mais automático, principalmente para usuários menos
experientes, uma vez que, este programa, necessita apenas das estruturas das
moléculas que serão utilizadas (ligante e alvo), e as coordenadas e dimensões
do sítio ativo. Outro motivo para a escolha do programa AutoDock Vina foi a
sua capacidade interna, já implementada, de utilizar os diversos núcleos de
processamento da máquina em seu algoritmo genético, dividindo o espaço de

36
busca de soluções entre os núcleos existentes para acelerar a realização do
docking molecular. Desta forma, o AutoDock Vina é mais rápido comparado
com outras soluções de docking, inclusive o próprio AutoDock.
4.2 Testes de desempenho computacional (“performance”)
Para a realização dos testes de desempenho, utilizou-se a infraestrutura
de um grid computacional montado no Laboratório de Química Teórica
Medicinal (LQTM). A máquina front-end, responsável direta por executar
efetivamente o programa GriDoMol para submeter os jobs para as demais
maquinas do ambiente de grid computacional, utiliza o processador Intel dual
core Xeon 2.80 GHz contendo 2GB de memoria RAM e executa o sistema
operacional Windows. As tasks (cálculos individuais de docking) foram
executadas em paralelo no ambiente de grid computacional homogêneo
(contendo oito máquinas), no qual cada máquina (contendo o componente
worker do OurGrid) possui dois processadores Intel Xeon e5410 quad core
2.33 GHz, com 16GB de memória RAM executando o sistema operacional
Linux Ubuntu 10.10. Todas as máquinas e o switch se comunicaram utilizando
interfaces de rede gigabit (1000Mb/s).
As análises quantitativas de tempo de processamento, por exemplo,
foram medidas com precisão em uma thread (um processamento contínuo
separado do programa principal) dedicada do programa GriDoMol. Esta thread
tem a função de analisar os arquivos recebidos por meio das máquinas
integrantes do grid computacional para o computador principal que está

37
executando o programa GriDoMol e, desta forma, controlar quais cálculos de
docking molecular foram finalizados ao receber seus respectivos resultados.
Para melhor observar o ganho de desempenho nos diferentes níveis de
paralelização dos cálculos realizados, definiram-se três principais métricas de
desempenho computacional. Estas métricas foram o Fator de divisão por Cores
(FdC), Fator de divisão por Instancias de execução (FdI) e o Fator de divisão
por Máquinas (FdM).
O fator de divisão por cores (FdC) é o coeficiente que determina o ganho
de desempenho adquirido, ao utilizar o nível de distribuição multicore, ou seja,
nos diversos núcleos de processamento das máquinas. Seu valor é
determinado pela divisão do tempo necessário para a realização dos cálculos
de docking molecular em máquinas que utilizem apenas um núcleo de
processamento, pelo tempo do mesmo número de máquinas com mais de um
núcleo de processamento sendo utilizado. Sua fórmula pode ser encontrada na
Equação 1.
FdC = T(MxC1)/T(MxCy) (1)
Nesta fórmula, o termo T(MxC1) é o tempo total gasto por “x” máquinas
que utilizem apenas um núcleo de processamento, enquanto o termo T(MxCy)
é o tempo total gasto pelas mesmas “x” máquinas, mas, desta vez, utilizando
“y” núcleos, para realizar a mesma tarefa. Por exemplo, o FdC previsto de um
cálculo que utiliza quatro máquinas e todos os oito núcleos de processamento
(cada) deveria ser igual a 8 (oito), uma vez que tende a ser idealmente 8 (oito)
vezes mais rápido do que o cálculo utilizando quatro máquinas onde apenas

38
um dos oito núcleos de processamento, em cada, está sendo utilizado. Esta
métrica é utilizada apenas no caso do programa AutoDock Vina, uma vez que,
através do parâmetro “cpu = y”, pode-se determinar quantos núcleos de
processamento serão utilizados nos cálculos de docking molecular com este
programa. É importante ressaltar que este recurso multicore do programa
AutoDock Vina é uma opção interna deste programa, tendo sido implementada
por seu desenvolvedor original. Sendo assim, o ganho de desempenho
proveniente deste paralelismo multicore não é o foco central deste trabalho de
mestrado, que enfatiza primariamente o ganho de desempenho devido à
paralelização das tarefas em um ambiente de grid computacional.
No programa AutoDock, usou-se a estratégia de enviar múltiplas
instâncias de execução deste programa para cada máquina, na tentativa de
utilizar a disponibilidade de núcleos de processamento ociosos para a
realização paralela dos cálculos individuais de docking molecular. Tal
abordagem está relacionada ao fato de que, diferentemente do programa
AutoDock Vina, o programa AutoDock não tem suporte a execução de forma
paralela entre os núcleos de processamento, sendo esta uma característica
interna, proveniente da sua implementação original, por parte dos autores e
desenvolvedores do mesmo. Uma vez que não há como controlar o fato de que
os cálculos de docking molecular simultâneos com o programa AutoDock,
realizados na mesma máquina, irão sempre ser distribuídos de forma
homogênea entre os núcleos de processamento, optou-se por utilizar uma
métrica diferente do FdC para medir este ganho de desempenho. Para medir,
portanto, o ganho de desempenho ao aumentar o número de instâncias, ou

39
seja, o número de cópias sendo executadas em paralelo na mesma máquina,
criou-se o conceito de Fator de divisão por Instancias de execução (FdI). A
fórmula desta métrica, FdI, é idêntica ao FdC e esta nomenclatura diferenciada
tem como objetivo diferenciá-lo do FdC, onde neste último, temos a segurança
de determinar (com o comando “cpu = y”), quantos núcleos estarão sendo
utilizados nos cálculos, em cada máquina utilizada no ambiente de grid
computacional.
O Fator de divisão por Máquinas (FdM) busca quantificar o ganho de
performance obtido na arquitetura distribuída do grid computacional. É
calculado segundo a Equação 2.
FdM = T(M1Cx)/T(MyCx) (2)
O termo T(M1Cx) indica o tempo total gasto por uma máquina utilizando
“x” núcleos de processamento, e o termo T(MyCx) é o tempo total gasto, para
executar a mesma tarefa, usando-se “y” máquinas e os mesmos “x” núcleos de
processamento do termo anterior. Utilizando exemplo parecido para ilustrar o
conceito de FdC, o FdM previsto, ao utilizar quatro máquinas e oito núcleos de
processamento cada, deveria, em tese, ser igual a 4 (quatro), uma vez que é
previsto que seja 4 (quatro) vezes mais rápido quando comparado com o
cálculo utilizando apenas uma máquina com os mesmos oito núcleos de
processamento, no caso do escalonamento de performance no grid ser
observado como essencialmente linear.
O conjunto de moléculas escolhido para os testes já está bem
estabelecido na literatura e já foi utilizado pelo grupo de quimiogenômica

40
estrutural da universidade de Estrasburgo (KELLENBERGER et al., 2004), em
um estudo comparativo. Um conjunto de 50 (cinquenta) receptores e seus
respectivos ligantes foi selecionado para a realização de re-docking (ver Tabela
2). O re-docking acontece quando se retira o ligante que está co-cristalizado
junto com o seu respectivo alvo biológico, e o programa de docking molecular
tenta reinserí-lo na macromolécula. Esta abordagem é muito utilizada para fins
de avaliação da capacidade preditiva dos programas de docking molecular.
Para obter um volume maior de demanda computacional e possibilitar melhores
comparações de desempenho, cada task (cálculo individual de docking
molecular em um complexo ligante+receptor) foi repetida 10 (dez) vezes,
totalizando um job contendo 500 (50 complexos x 10 cópias cada) tarefas
(tasks).
A preparação das moléculas, como a retirada das moléculas de água,
adição dos átomos de hidrogênio faltantes e adição das cargas dos átomos, foi
realizada através do uso da ferramenta computacional AutoDock Tools (ADT,
2012). Os arquivos grid parameter file (gpf) e docking parameter file (dpf),
utilizados pelo AutoGrid e pelo AutoDock, respectivamente, foram gerados
automaticamente através dos scripts disponibilizados pelo pacote AutoDock
Tools. Esta etapa inicial de preparação das moléculas é obrigatória para a
realização dos cálculos de docking molecular nos programas AutoDock e
AutoDock Vina.
Além dos parâmetros padrões necessários para a realização dos
cálculos de docking em si, outros parâmetros foram utilizados para melhorar os
resultados obtidos, e também para tentar padronizar o tempo que cada task

41
(cálculo individual de docking) é realizada e, assim, viabilizar a reprodução dos
testes de desempenho.
Tabela 2 – Lista do conjunto de complexos ligante-receptor selecionados para este estudo.
*N = Número de ligações com rotação livre. MW = Massa Molar do ligante, em g/mol. R = Resolução, em
Ångstrons. ID = identidade do respectivo código PDB.
# ID R RECEPTOR LIGAND MW N 1 1abe 1.7 L-Arabinose-Binding Protein L-Arabinose 150.1 4 2 1acj 2.8 Acetylcholinesterase Tacrine 198.3 1 3 1acm 2.8 Aspartate Carbamoyltransferase N-Phosphonacetyl-L-Aspartate 251.1 6 4 1aha 2.2 alpha-Momorcharin Adenine 135.1 1 5 1baf 2.9 Anti-dinitrophenyl-spin-label monoclonal
antibody Fab fragment 1-Hydroxy-2,2,6,6,-Tetramethyl-4-(2-4,Dinitro-
5-(Methyl-Aminoethyl-Amino)Phenylamino)PIPERIDINE
396.5 12
6 1cbs 1.8 Cellular Retinoic-Acid-Binding Protein Type II
Retinoic Acid 299.4 10
7 1com 2.2 Chorismate Mutase Prephenate 224.2 5 8 1dbb 2.7 Fab' Fragment Of The Db3 Anti-Steroid
Monoclonal Antibody Progesterone 314.5 4
9 1eap 2.5 Catalytic Antibody 17E8 Phenyl [1-(1-N-Succinylamino)Pentyl] Phosphonate
341.3 11
10 1eed 2.0 Endothiapepsin Cyclohexyl Renin Inhibitor Pd125754 299.4 10 11 1fki 2.2 Fk506 Binding Protein Rotamase Inhibitor (21S)-1-Aza-4,4-Dimethyl-
6,19-Dioxa-2,3,7,20-Tetraoxobicyclo Pentacosane
437.6 1
12 1hri 3.0 Human Rhinovirus 14 1-[6-(2-Chloro-4-Methyxyphenoxy)-Hexyl]-Imidazole
308.8 10
13 1hsl 1.8 Histidine-Binding Protein L-Histidine 155.2 4 14 1igj 2.5 Fab (Igg2A, kappa) Digoxin 520.7 10 15 1ive 2.4 Influenza A Subtype N2 Neuraminidase 4-(Acetylamino)-3-Aminobenzoic Acid 193.2 4 16 1lah 2.0 Lysine, Arginine, Ornithine-Binding Protein Ornithine 133.2 6 17 1lst 1.8 Lysine-, Arginine-, Ornithine-Binding Protein Lysine 146.2 7 18 1mcr 2.7 Immunoglobulin delta Light Chain Dimer N-Acetyl-L-His-D-Pro-Oh 293.3 6 19 1mrg 1.8 alpha-Momorcharin Adenine 135.1 1 20 1mrk 1.6 alpha-Trichosanthin Formycin 267.3 6 21 1mup 2.4 Major Urinary Protein Complex 2-(Sec-Butyl) Thiazoline 143.3 4 22 1rob 1.6 Ribonuclease A Cytidylic Acid 321.2 5 23 1snc 1.6 Staphylococcal Nuclease 3-Prime,5-Prime-Deoxythymidine
Bisphosphate 398.2 3
24 1srj 1.8 Streptavidin Naphthyl-Haba 291.3 4 25 1stp 2.6 Streptavidin Biotin 243.3 3 26 1tdb 2.6 Thymidylate Synthase 5-Fluoro-2'-Deoxyuridine-5'-Monophosphate 324.2 0 27 1tng 1.8 Trypsin Aminomethylcyclohexane 114.2 2 28 1tnl 1.9 Trypsin Tranylcypromine 133.2 2 29 1tph 1.8 Triosephosphate Isomerase Phosphoglycolohydroxamate 169.0 4 30 1tpp 1.4 beta-Trypsin P-Amidino-Phenyl-Pyruvate 206.2 6 31 1ukz 1.9 Uridylate Kinase Adenosine-5'-Diphosphate 345.2 6 32 1ulb 2.7 Purine Nucleoside Phosphorylase Guanine 151.1 1
33 2ack 2.4 Acetylcholinesterase Edrophonium 166.3 6 34 2ak3 1.8 Adenylate Kinase Isoenzyme-3 Adenosine Monophosphate 345.2 7
35 2cgr 2.2 Igg2B (kappa) Fab Fragment Antigen N-(P-Cyanophenyl)-N'-(Diphenylemethyl)Guanidineacetic Acid
383.4 7
36 2cht 2.2 Chorismate Mutase Endo-Oxabicyclic Inhibitor 226.2 3 37 2cmd 1.8 Malate Dehydrogenase Citric Acid 189.1 6 38 2dbl 2.9 Fab' Fragment Of Monoclonal Antibody Db3 5-alpha-Pregnane-3-beta-Ol-Hemisuccinate 417.6 9 39 2gbp 1.9 D-Galactose D-Glucose Binding Protein Glucose 180.2 6 40 2r07 3.0 Rhinovirus 14 Antiviral Agent WIN VII 314.4 9 41 2sim 1.6 Sialidase 2-Deoxy-2,3-Dehydro-N-Acetyl-Neuramic 290.3 10 42 3hvt 2.9 Reverse Transcriptase Dipyridodiazepinone Nevirapine 266.3 1 43 3ptb 1.7 beta-Trypsin Benzyldiamine 121.2 1 44 3tpi 1.9 Trypsinogen Pancreatic Trypsin Inhibitor 230.3 11 45 4cts 2.9 Citrate Synthase Oxaloacetate ion 130.1 3 46 4fab 2.7 4-4-20 (IgG2A) Fab Fragment - Fluorescein
(Dianion) Complex 2-methyl-2,4-pentanediol 411.2 4
47 6abp 1.6 L-Arabinose-Binding Protein L-Arabinose 148.1 4 48 7tim 1.9 Triosephosphate Isomerase Phosphoglycolohydroxamate 169.0 4 49 8atc 2.5 Aspartate Carbamoyltransferase N-Phosphonacetyl-L-Aspartate 251.1 6 50 8gch 1.6 gamma-Chymotrypsin Gly-Ala-Trp 332.4 9

42
4.2.1 Programa AutoDock
No programa AutoDock, pelo fato de sua busca ser tipicamente robusta
o suficiente, apenas o parâmetro seed (semente inicial para o gerador de
números aleatórios) foi ajustado com um valor fixo (número = 495829104), a
fim de permitir a reprodutibilidade dos procedimentos de cálculo. Este valor foi
atribuído arbitrariamente, utilizando um dos valores gerados automaticamente
em uma execução a priori da realização dos testes. Para compensar a falta de
paralelismo a nível multicore para o programa AutoDock, decidiu-se escalonar
a utilização dos núcleos de processamento através do envio de mais de uma
instância de execução do programa AutoDock em cada máquina, quando
pertinente.
Devido aos mapas gerados pelo programa AutoGrid serem grandes, em
termos de espaço ocupado em disco, usou-se a estratégia de comprimir os
mapas em um arquivo (formato zip) antes de enviar os resultados para a
máquina front-end e, desta forma, reduzir o tempo de transferência de
arquivos.
Tanto para o AutoGrid, quanto para o AutoDock, foi necessária a
criação de rotinas computacionais (scripts), para permitir a compressão,
extração (no caso do AutoDock) dos mapas atômicos e para efetuar a
execução paralela (nos casos onde mais de uma instância de cálculo com o
programa AutoDock é enviado para a mesma máquina) como meio de driblar a
limitação apresentada pelo OurGrid, que limita cada tarefa (task) a uma única
linha de comando de execução.

43
Vale ressaltar que, como cada tarefa contendo cálculos em paralelo do
programa AutoDock é contada como uma única tarefa pelo ambiente de grid
computacional OurGrid, a tarefa, em cada máquina, só será concluída quando
todas as execuções distribuídas nas múltiplas instâncias do programa
AutoDock finalizarem.
4.2.2 Programa AutoDock Vina
No AutoDock Vina, o valor do parâmetro exhaustiveness, responsável
por indicar o tempo gasto em buscas por um mínimo global em cada cálculo de
docking, foi configurado para o valor igual a 16 (dezesseis), que representa o
dobro do valor padrão (oito). Também foi modificado o valor do parâmetro
seed, que indica a semente inicial para o gerador de números aleatórios, por
onde o algoritmo genético começa sua busca, tornando-o fixo (número =
495829104) ao invés de aleatório. Este valor foi atribuído arbitrariamente,
utilizando um dos valores gerados automaticamente em uma execução a priori
da realização dos testes. Assim, tentou-se padronizar o processo de execução
de cada cálculo de docking, com o intuito de tornar o procedimento
reprodutível, uma vez que o algoritmo irá sempre começar pelo mesmo lugar.
Durante os testes, para determinar o impacto da quantidade de
máquinas no desempenho do grid computacional, foi alternada a quantidade de
computadores envolvidos no dimensionamento do grid computacional, variando
entre 1 e 8 máquinas em funcionamento para o AutoDock Vina. O
escalonamento das máquinas para o AutoDock variou entre 1 e 6,
sistematicamente.

44
Também houve variação no número de núcleos de processamento
utilizados para determinar o impacto multicore, no caso do programa AutoDock
vina, e de múltiplas execuções ou instâncias paralelas, no caso do AutoDock.
Como o foco deste trabalho é a paralelização em grid computacional, ou seja, a
distribuição dos cálculos em grande número de máquinas, e para abreviar a
quantidade de cálculos, a progressão para o número de núcleos de
processamento (cores) utilizados foi feita de forma geométrica, variando-se em
1, 2, 4 e 8 núcleos de processamento.

45
5 RESULTADOS E DISCUSSÕES
5.1 O programa GriDoMol
O programa GriDoMol, desenvolvido neste trabalho, fornece uma
interface simples e intuitiva, em conjunto com funcionalidades que permitem
criar, enviar e gerenciar cálculos de docking molecular em grande escala, no
ambiente distribuído de um grid computacional. A descrição quantitativa do
código fonte desenvolvido durante a realização deste trabalho pode ser
encontrada na Tabela 3. Para obter estes custos estimados usou-se o
programa SLOCCount (SLOCCount, 2012). Este programa tem sido utilizado
para uma avaliação mais quantitativa do esforço e custo estimados no
desenvolvimento de diversos softwares, inclusive, recentemente, aplicado a
análise do núcleo (Kernel) do sistema operacional Linux (The Linux Kernel: It’s
Worth more!, 2012). As estimativas realizadas pelo programa SLOCCount leva
em consideração o modelo Construtive Cost Model (COCOMO). Uma
descrição detalhada do modelo COCOMO e como ele é utilizado pelo
programa SLOCCount em suas estimativas pode ser encontrado no guia do
usuário (SLOCCount User’s Guide, 2013). Vale ressaltar que um dos
resultados provenientes da análise do código GriDoMol, feito pelo SLOCCount,
foi o “esforço estimado para desenvolvimento" (“Development Effort Estimate”),
com um valor de 0,58 pessoas/ano, o que significa praticamente 1 (uma)
pessoa em 2 (dois) anos, que coincide basicamente com o período (de dois
anos) do projeto de mestrado.

46
Tabela 3 – Características do código fonte implementado no programa GriDoMol obtido a partir
do programa SLOCCount (SLOCCount, 2012).
Número de desenvolvedores
(estimado)
Esforço estimado para
desenvolvimento (pessoas/ano)
Linhas de
Código
1,33 0,58 2.756
Com o intuito de apresentar as informações importantes para
acompanhar o andamento das tarefas executadas no ambiente de grid
computacional, a estrutura da tela principal (Figura 5.1) conta com os seguintes
campos: Info, Workers e as abas do Broker e do Peer.
No campo Info, informações gerais sobre o andamento das tarefas são
apresentadas, tal como a data e hora do inicio e fim do cálculo e a
porcentagem dos cálculos já concluídos.
Os detalhes específicos sobre cada tarefa agendada para execução no
ambiente de grid computacional são exibidos em uma tabela presente no
campo Workers. Esta tabela conta com as colunas task, worker, status e start
time.
A coluna task mostra o nome das moléculas envolvidas em cada cálculo
de docking molecular, utilizando para tal nomenclatura o formato “receptor-
ligante”. Na coluna worker, é exibido o nome da máquina responsável pelo
processamento do cálculo em questão. O status indica o estado em que cada
tarefa se encontra, dentre eles: não iniciado (unstarted), em execução
(running), encontrou uma falha (failed) e concluído (finished). Por último, a
coluna start time mostra o horário no qual houve o inicio de cada cálculo.

47
Figura 5.1 – Tela inicial do GriDoMol, sendo executado no sistema operacional Windows.
As abas Broker e Peer registram as atividades de cada um desses
componentes, exibindo informações de avisos, como por exemplo, o número
de máquinas disponíveis, as mensagens de erro e outras mensagens de
comunicação de cada um destes componentes do sistema.
A interface para a preparação (ver Figura 5.2) dos arquivos necessários
para os cálculos de docking no ambiente distribuído é acessada através da
opção new job, disponível no menu file da interface principal. Nesta tela, é
solicitado o preenchimento dos dados de entrada, de acordo com os campos
General Information, Ligand Information, Receptor Information e Molecules List,
de forma que:
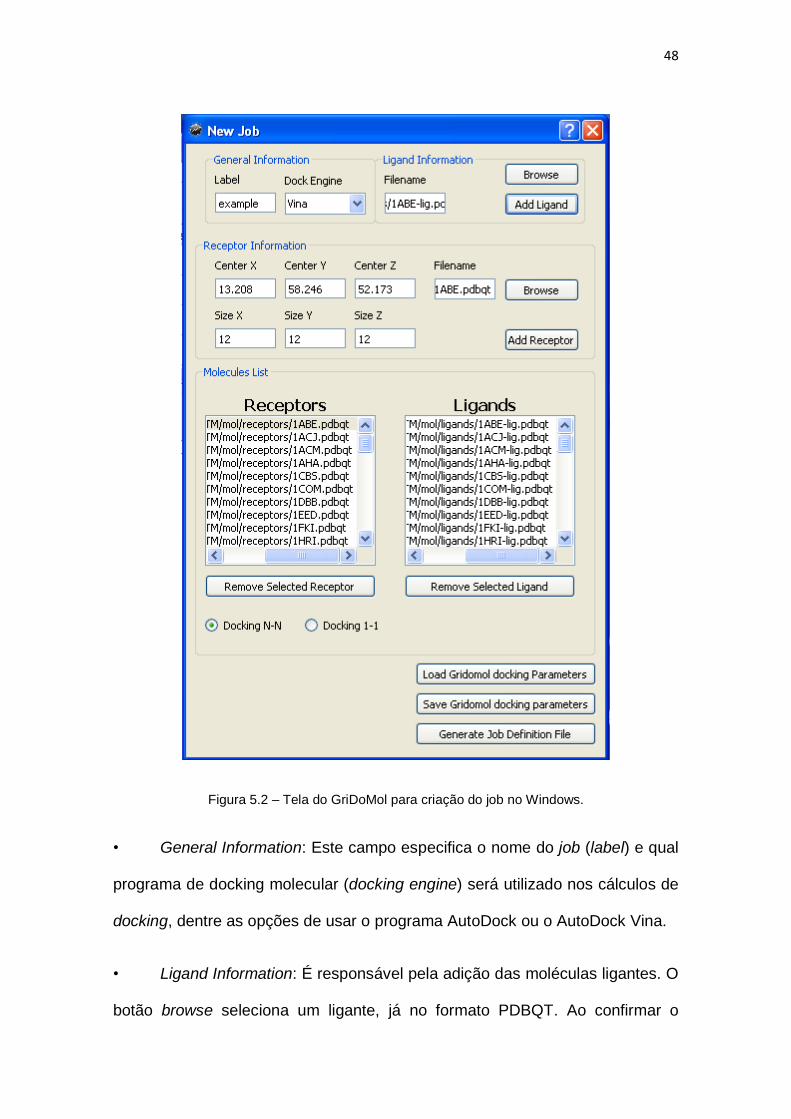
48
Figura 5.2 – Tela do GriDoMol para criação do job no Windows.
• General Information: Este campo especifica o nome do job (label) e qual
programa de docking molecular (docking engine) será utilizado nos cálculos de
docking, dentre as opções de usar o programa AutoDock ou o AutoDock Vina.
• Ligand Information: É responsável pela adição das moléculas ligantes. O
botão browse seleciona um ligante, já no formato PDBQT. Ao confirmar o

49
ligante, o botão add ligand serve para adicioná-lo ao campo molecules list, o
qual lista todas as moléculas escolhidas para os cálculos.
• Receptor Information: Adiciona os receptores de maneira análoga ao
processo de seleção dos ligantes. Além de informar o arquivo PDBQT do
receptor, é necessário preencher as coordenadas tridimensionais (x, y e z) do
centro de seu sitio ativo e a área de busca a partir do centro do sitio ativo,
sendo que estas informações podem ser obtidas através do uso de programas
com interface gráfica de visualização do alvo biológico, como por exemplo, o
AutoDock Tools, que é totalmente compatível com o pacote de programas do
AutoDock e AutoDock Vina.
• Molecules List: A lista de moléculas que farão parte do cálculo de
docking (ou virtual screening) é apresentada neste campo. Há a opção de
remover os receptores e ligantes indesejáveis através dos botões remove
selected receptor ou remove selected ligand, respectivamente. Ainda nesta
etapa, é determinado o tipo de cálculo de docking que será realizado, podendo-
se escolher entre os cálculos de docking N-N ou os cálculos de docking 1-1. Na
modalidade docking N-N, o docking de todos os ligantes será realizado com
todos os receptores, no que costuma ser chamado de cross-docking. Já na
modalidade docking 1-1, cada receptor estará envolvido apenas no cálculo com
o ligante que estiver na mesma sequência (numeração linear) da lista.
Com o intuito de facilitar uma futura reutilização e alteração do conjunto
de moléculas selecionadas para os cálculos e seus parâmetros, foram
adicionadas ao GriDoMol as funcionalidade de salvar e carregar parâmetros de

50
cálculos de docking, através dos botões save GriDoMol docking parameters e
load GriDoMol docking parameters, respectivamente. Ao se carregar o
GriDoMol setup file (gsf), é possível realizar alterações de dados como:
adicionar e/ou remover moléculas, modificar as coordenadas da localização do
sitio ativo, o tamanho da área de busca em torno do sitio ativo, e re-selecionar
o programa de docking que irá realizar o cálculo.
Após a adição dos dados necessários, a função generate job definition
file cria o arquivo job description file (jdf) necessário para a realização do
cálculo de docking no ambiente de grid computacional. O job description file,
para a execução do módulo AutoGrid, responsável por gerar os mapas
atômicos de cada receptor utilizado no cálculo de docking com o programa
AutoDock, é criado, automaticamente, junto com o arquivo jdf do módulo
AutoDock, quando o dock engine selecionado é o AutoDock.
A Figura 5.3 mostra o esquema de comunicação entre o programa
GriDoMol e os demais programas utilizados neste trabalho. O Fluxo de
execução começa pela preparação inicial das moléculas que serão utilizadas,
como, por exemplo, adicionar átomos de hidrogênio, remover as moléculas de
água e converter os arquivos contendo as estruturas moleculares para o
formato PDBQT. Após esta preparação inicial, o usuário pode, através do
GriDoMol: i) Configurar o grupo de máquinas que farão parte do ambiente de
grid computacional, utilizando o arquivo Site Description File (SDF) do
componente Peer; ii) Criar ou carregar os aqruivos GriDoMol Setup File (GSF)
contendo as informações sobre as moléculas que serão utilizadas; iii) Criar o
arquivo Job Description File (JDF), utilizado para realizar os cálculos de

51
docking molecular de forma distribuída, e submete-lo para o componente
Broker.
Após enviar o job (conjunto de cálculos de docking molecular) para o
component Broker, o mesmo se encarrega de distribuir as tasks (cálculo
individual de docking) de forma otimizada entre os componentes Worker
presente em cada máquina encarregada de realizar cálculos de docking no
ambiente de grid computacional. O componente Peer analisa a disponibilidade
das máquinas em tempo real, e informa o componente Broker caso alguma
máquina fique indisponível. Cada máquina que executa o componente Worker
realiza cálculos de docking utilizando os programas AutoDock e AutoDock
Vina. Quando o cálculo de docking termina, os resultados são enviados para a
máquina do usuário.
Para determinar o poder computacional do ambitente de grid
computacional, controlado pelo GriDoMol, em FLOPS (operações de ponto
flutuante por segundo) utilizou-se neste estudo a ferramenta Linpack (Linpack,
2013). A ferramenta Linpack é muito utilizada para determinar o poder
computacional, incluindo testes de desempenho de supercomputadores (Top
500 Supercomputers, 2012). O resultado obtido para uma das máquinas
usadas neste trabalho, em nosso laboratório, foi de 56,8 GFLOPS (109 FLOPS)
e, como as máquinas que compõe o ambiente de grid computacional deste
presente trabalho são homogêneas, o poder computacional estimado de todo o
ambiente grid é aproximadamente 454,2 GFLOPS.

52
Fig
ura
5.3
- Esq
ue
ma
da
arq
uite
tura
de
co
mun
icaçã
o e
ntre
os s
oftw
are
s d
e c
ad
a m
áq
uin
a u
tiliza
do
s n
este
trab
alh
o.

53
5.2 Resultados dos cálculos de docking
O total de cálculos realizados pode ser observado na tabela 4. Além dos
testes de desempenho realizados utilizando os programas AutoDock e
AutoDock Vina, também realizou-se testes adicionais para investigar alguns
comportamentos observados no ambiente de grid computacional. Os testes de
ordenamento dos cálculos de docking dentro de um job foram realizados para
tentar determinar se a ordem em que os cálculos de docking são realizados
influência o tempo total de execução do job no ambiente de grid computacional.
Já os testes de trasferência de arquivos foram realizados para tentar
determinar o impacto da etapa de transferência de arquivos no tempo total do
job.
Tabela 4 – Total de cálculos realizados.
Teste jobs Cálculos individuais
AutoDock 24 12.000
AutoDock Vina
32 16.000
Testes de ordenamento
40 20.000
Testes de transferência
8 4.000
Total 104 52.000
Antes do inicio de cada cálculo de docking, observou-se um atraso
(delay) típico de aproximadamente 10 segundos entre o comando de envio de
cada cálculo (task) na interface do programa GriDoMol, e a realização efetiva

54
do mesmo na máquina de trabalho (worker). Este delay é típico da arquitetura
de grid computacional e se deve normalmente as comunicações entre os
componentes do grid computacional. Estas comunicações incluem verificações
de disponibilidade de máquinas, ou seja, se elas ainda estão ligadas e
conectadas ao ambiente de grid computacional, e verificação da presença dos
requisitos mínimos necessários para a realização da tarefa, na máquina
escolhida pelo grid computacional.
Vale ressaltar que, devido às comunicações e transferência de arquivos
no ambiente de grid computacional, os cálculos com apenas uma máquina são
mais lentos do que se fossem realizados fora do ambiente de grid
computacional. Utilizando, por exemplo, rotinas computacionais como shell
scripts ou arquivos de lote (batch), é possível realizar estes cálculos que
utilizam apenas uma máquina numa fração do tempo realizado no ambiente de
grid computacional. Contudo, como o propósito central do ambiente de grid
computacional é a utilização de processamento distribuído em varias
máquinas, esta situação de execução em apenas uma máquina é bastante
improvável. Em adição, vale ressaltar que mesmo que o tempo de execução do
job (conjunto de cálculos) no grid computacional operando com apenas 1 (uma)
máquina fosse inferior à utilização de shell scripts ou arquivos de lote para
executar todas estas tarefas (cálculos individuais de docking) apenas nesta
máquina, ainda assim a interface gráfica do GriDoMol e o gerenciamento
sequencial das tarefas individuais facilitaria bastante o processo de cálculo,
sem a necessidade de nenhum recurso adicional (shell scripts ou arquivos de
lote).
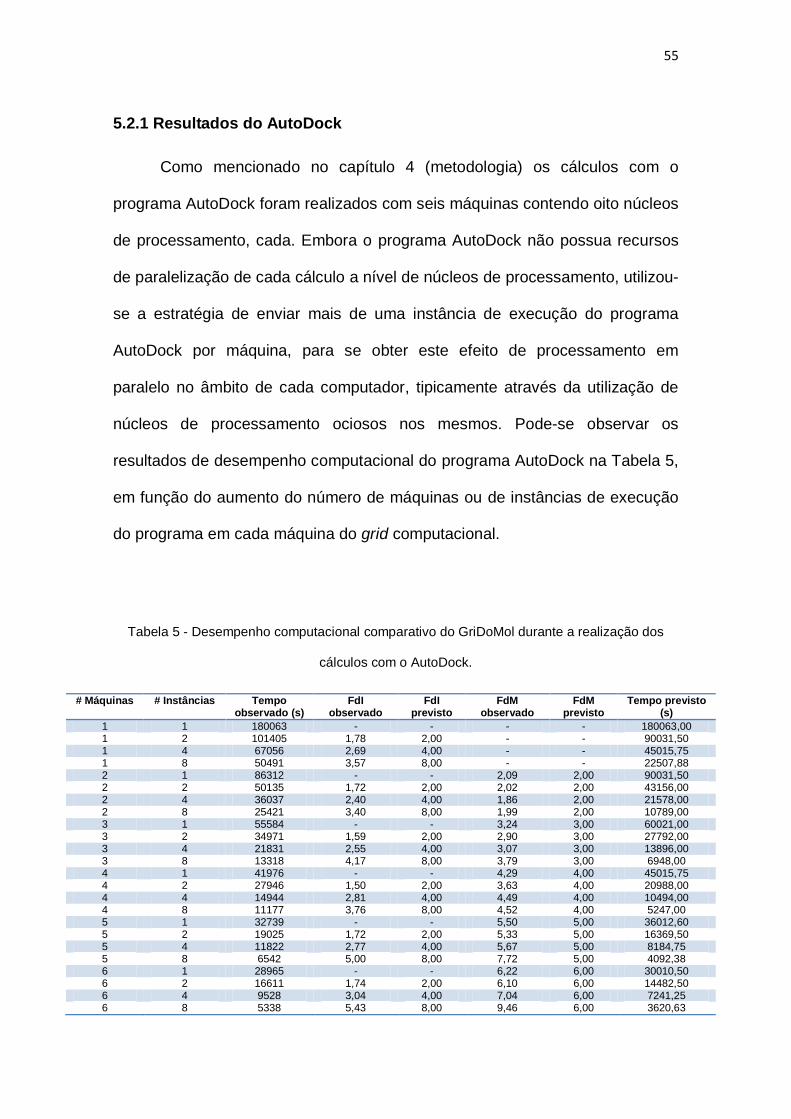
55
5.2.1 Resultados do AutoDock
Como mencionado no capítulo 4 (metodologia) os cálculos com o
programa AutoDock foram realizados com seis máquinas contendo oito núcleos
de processamento, cada. Embora o programa AutoDock não possua recursos
de paralelização de cada cálculo a nível de núcleos de processamento, utilizou-
se a estratégia de enviar mais de uma instância de execução do programa
AutoDock por máquina, para se obter este efeito de processamento em
paralelo no âmbito de cada computador, tipicamente através da utilização de
núcleos de processamento ociosos nos mesmos. Pode-se observar os
resultados de desempenho computacional do programa AutoDock na Tabela 5,
em função do aumento do número de máquinas ou de instâncias de execução
do programa em cada máquina do grid computacional.
Tabela 5 - Desempenho computacional comparativo do GriDoMol durante a realização dos
cálculos com o AutoDock.
# Máquinas # Instâncias Tempo observado (s)
FdI observado
FdI previsto
FdM observado
FdM previsto
Tempo previsto (s)
1 1 180063 - - - - 180063,00 1 2 101405 1,78 2,00 - - 90031,50 1 4 67056 2,69 4,00 - - 45015,75 1 8 50491 3,57 8,00 - - 22507,88 2 1 86312 - - 2,09 2,00 90031,50 2 2 50135 1,72 2,00 2,02 2,00 43156,00 2 4 36037 2,40 4,00 1,86 2,00 21578,00 2 8 25421 3,40 8,00 1,99 2,00 10789,00 3 1 55584 - - 3,24 3,00 60021,00 3 2 34971 1,59 2,00 2,90 3,00 27792,00 3 4 21831 2,55 4,00 3,07 3,00 13896,00 3 8 13318 4,17 8,00 3,79 3,00 6948,00 4 1 41976 - - 4,29 4,00 45015,75 4 2 27946 1,50 2,00 3,63 4,00 20988,00 4 4 14944 2,81 4,00 4,49 4,00 10494,00 4 8 11177 3,76 8,00 4,52 4,00 5247,00 5 1 32739 - - 5,50 5,00 36012,60 5 2 19025 1,72 2,00 5,33 5,00 16369,50 5 4 11822 2,77 4,00 5,67 5,00 8184,75 5 8 6542 5,00 8,00 7,72 5,00 4092,38 6 1 28965 - - 6,22 6,00 30010,50 6 2 16611 1,74 2,00 6,10 6,00 14482,50 6 4 9528 3,04 4,00 7,04 6,00 7241,25 6 8 5338 5,43 8,00 9,46 6,00 3620,63

56
Figura 5.4 - Gráfico de desempenho do AutoDock em função do tempo. As curvas com a
legenda no formato Ex (onde x se refere ao número de núcleos de processamento utilizados)
se refere aos valores observados enquanto as curvas no formato Exp representam os valores
previstos.
Nota-se que, ao aumentar o número de máquinas envolvidas na
execução dos cálculos de docking, o tempo de execução dos 500 cálculos de
docking se aproxima do tempo previsto (Figura 5.4). Isto deve ocorrer devido
ao aumento do número de transferências simultâneas de arquivos, observadas
ao se utilizar mais máquinas nos cálculos de docking no ambiente de grid
computacional. Estas transferências simultâneas reduzem o tempo total gasto
com transferências, e consequentemente o tempo total de execução. Por
exemplo, ao utilizar apenas uma máquina, as transferências são,
obrigatoriamente, sequenciais enquanto que, ao utilizar mais de uma máquina,

57
ocorre a paralelização fortuita destas transferências de forma que isto aumenta
o desempenho computacional dos cálculos do conjunto de moléculas como um
todo. Como o tamanho médio dos arquivos transferidos para a realização dos
cálculos no Autodock, em média, está entre 1MB a 3MB (já comprimido no
formato zip), este efeito se torna significativo para este programa, porém
desprezível para o Autodock Vina, onde o tamanho dos arquivos transferidos é
bem menor (entre 0.1MB e 0.7MB, tipicamente).
Como se pode notar no gráfico do FdI observado durante a utilização do
programa AutoDock (Figura 5.5), o desempenho ao aumentar o número de
instâncias de execução do programa AutoDock ficou aquém do previsto. Como
visto anteriormente, cada tarefa contendo cálculos em paralelo do AutoDock é
contada como uma única tarefa pelo ambiente de grid computacional. O tempo
total de execução de cada um destes pequenos conjuntos de instâncias do
programa que é executado em cada máquina é sempre o tempo que o cálculo
mais demorado deste conjunto leva para ser concluído. Nesta situação, onde
todas as instâncias paralelas de execução do cálculo já terminaram, restando
apenas uma executando em apenas um núcleo, deve haver um grande número
de núcleos de processamento ociosos, esperando o último cálculo do conjunto
ser concluído no último núcleo ativo (não ocioso) de cada máquina. Esta
ociosidade dos núcleos de processamento compromete o tempo total de
execução, uma vez que estes núcleos ociosos poderiam estar realizando os
cálculos que ainda precisam ser feitos na lista de tasks do job (lista completa
de cálculos de docking a ser realizados). Este problema não pode ser

58
contornado, uma vez que, para o OurGrid, cada tarefa contendo multiplas
instâncias de execução é vista como uma única tarefa.
Figura 5.5 - Gráfico do FdI para o programa AutoDock. As legenda no formato Mx (onde x se
refere ao número de máquinas utilizadas) mostra os valores observados em relação ao
previsto.
Vale ressaltar que o baixo desempenho observado no FdI,
especialmente nos casos com oito instâncias de execução, pode ter sido
causado pela sobrecarga de demanda computacional na máquina, deixando
pouco processamento para as atividades essenciais de gerenciamento do
sistema operacional da máquina. Além disto, parte deste baixo desempenho se
deve ao fato de que o programa AutoDock não foi desenvolvido para ser
executado em múltiplas instâncias de execução e algumas operações deste

59
programa, como, por exemplo, o excesso de escrita em disco, observado em
maior escala no estudo do Dovis (JIANG et al., 2008), apresentado na seção
2.4 deste trabalho, pode ter contribuído para o baixo valor de FdI observado em
relação ao esperado (Tabela 5).
Figura 5.6 – Gráfico do FdM para o programa AutoDock. As legenda no formato Ex (onde x se
refere ao número de instâncias de execução utilizadas) mostra os valores observados em
relação ao previsto.
O aumento de desempenho no programa AutoDock no ambiente de grid
computacional, conforme pode-se observar no gráfico a Figura 5.6, fica
próximo do previsto. Por conta do tamanho e quantidade de arquivos enviados
para a realização de cálculos de docking no programa AutoDock, a etapa de

60
transferência de arquivos é mais longa. Este efeito torna o cálculo base com
apenas uma máquina, muito mais demorado. Ao aumentar o número de
máquinas, além do ganho de desempenho através da distribuição de
processamento computacional dos cálculos de docking entre as máquinas do
ambiente de grid computacional, as transferências de dados aceleraram ainda
mais o desempenho, uma vez que o tamanho dos arquivos para realização de
múltiplos cálculos de docking no programa AutoDock são consideravelmente
grandes. Além disto, como visto na analise do FdI, nos exemplos com oito
instâncias de execução do Autodock, deve haver um elevado número de
núcleos de processamento parados esperando pelo último cálculo de cada
pacote de cálculos ser concluído, causando uma ociosidade de núcleos de
processamento que poderiam já estar sendo utilizados para a realização das
próximas tarefas individuais (cálculos de docking). Ao aumentar o número de
máquinas, ocorre a simultaneidade da ociosidade dos núcleos de
processamento, fazendo com que os cálculos sejam mais rápidos comparados
com a ociosidade sequencial e aditiva do exemplo com uma máquina. Por
exemplo, em um conjunto de cálculos realizados em quatro máquinas, há uma
chance de que estes núcleos de processamento possam estar
simultaneamente ociosos, a espera do ultimo cálculo ser concluído no último
núcleo ativo em cada máquina, e com isso, este tempo ocioso simultâneo é
contado uma única vez. Desta forma, acredita-se que esta ociosidade
simultânea fez com que os cálculos com mais máquinas fossem mais rápido do
que o previsto, uma vez que o FdM previsto é calculado tendo como base os
cálculos com apenas uma máquina, onde esta ociosidade é sequencial. Este

61
efeito pode ser melhor observado nos cálculos com oito instâncias de execução
(E8) nos casos com cinco e seis máquinas.
Tabela 6 – Tempo total estimado de comunicação entre os componentes do ambiente de grid
computacional.
Número de cálculos individuais de
Docking
Instâncias de execução do
programa AutoDock
Número de tasks
Tempo total de comunicação no ambiente de grid
(segundos)
500 1 500 5000
500 2 250 2500
500 4 125 1250
500 8 63 630
Como visto anteriormente (seção 5.2), observou-se um atraso (delay)
típico de aproximadamente 10 segundos entre o comando de envio da tarefa
(task) e a realização efetiva desta tarefa na máquina de trabalho (worker). Este
delay ocorre antes da execução de cada uma das tarefas (tasks) realizadas no
ambiente de grid computacional. No caso do programa AutoDock, ao aumentar
o número de instâncias de execução do programa em cada máquina, também
houve uma diminuição do número de tarefas (tasks), uma vez que, com
múltiplas instâncias de execução, cada task contém mais de um cálculo de
docking. Desta forma, ao aumentar o número de instâncias de execução do
programa AutoDock em cada máquina, além de diminuir o número de
processadores ociosos em cada máquina (como se observa na analise do FdI),
o tempo total de comunicação também é reduzido entre os componentes do
ambiente de grid computacional, comparado com o cálculo que utiliza apenas
uma instância de execução do programa AutoDock em cada máquina. Por

62
exemplo, o tempo total de comunicação (630 segundos) em cada job que utiliza
oito instâncias de execução do programa AutoDock foi bem menor do que o
tempo de comunicação (5000 segundos) observado nos jobs com apenas uma
instância de execução do programa AutoDock, conforme pode ser observado
na Tabela 6.
O aumento do número de máquinas utilizadas na realização dos cálculos
de docking também reduz o tempo total de comunicação entre os componentes
do ambiente de grid computacional, comparado com o tempo total de
comunicação sequencial em cálculos utilizando apenas uma máquina. Isto
ocorre, devido a simultaneidade destas comunicações que podem ocorrer
fortuitamente ao longo da execução do job, Esta simultaneidade das
comunicações entre as máquinas ocorre, obrigatoriamente, pelo menos no
primeiro conjunto de tarefas (tasks) enviado ao ambiente de grid
computacional, para múltiplas máquinas.
Ao examinar todos os testes do GriDoMol com o programa AutoDock,
pode-se observar que ao utilizar mais máquinas, ao invés de mais instâncias
de execução, o processo mostrou-se mais eficiente, levando menor quantidade
total de tempo. Por exemplo, utilizar quatro máquinas e apenas uma instância
de execução (ver Tabela 5 - tempo de execução: 41.976 segundos) é mais
rápido do que utilizar uma máquina e quatro instâncias de execução (ver
Tabela 5 - tempo de execução: 67.056 segundos). Ao utilizar ambas as formas
de distribuição para a realização dos cálculos de docking, ambiente de grid
computacional e múltiplas instâncias de execução do AutoDock, obteve-se uma
redução do tempo total de execução para apenas 3% (tempo de execução:

63
5.338 segundos) do tempo necessário comparado com o cálculo não
paralelizado (tempo de execução: 180.063 segundos), ou seja, feito com
apenas uma máquina e uma instância de execução do programa AutoDock.
Desta forma, ao utilizar estas formas de distribuição, os cálculos foram
realizados com o desempenho 33,73 vezes mais rápido, no ambiente de grid
computacional acessado pelo GriDoMol.
5.2.1.1 Reprodutibilidade do tempo de execução do AutoDock
Com o intuito de determinar se os parâmetros utilizados nos cálculos de
docking com o programa AutoDock os tornaram reprodutíveis em demanda
computacional, o tempo de execução de cada um destes cálculos para os
casos de uma máquina com uma instância de execução (Tabela 7), uma
máquina com oito instâncias de execução (Tabela 8) e seis máquinas com uma
instância de execução (Tabela 9), foram analisadas. Esta escolha de números
de máquinas e instâncias de execução do programa AutoDock utilizados neste
teste foi feita com o intuito de analisar possíveis impactos no tempo de
execução individual de cada cálculo ao aumentar o número de máquinas ou de
instâncias de execução simultânea do programa AutoDock. Os dados de tempo
utilizados nesta análise foram obtidos através dos arquivos de saída dos
cálculos individuais.
Observa-se nestas tabelas (Tabela 7, 8 e 9) que os valores obtidos em
cada cópia do cálculo se aproxima da média e que o desvio padrão é baixo. O
tempo de execução individual de cada cálculo no conjunto de cálculos com oito

64
instâncias de execução do programa AutoDock (Tabela 8) apresenta, em geral,
uma média mais alta que seus análogos com apenas uma instância de
execução do programa AutoDock (Tabela 7 e 9). Isto pode ter ocorrido devido a
possível sobrecarga dos núcleos de processamento ao utilizar todos os oito
núcleos de processamento de cada máquina, uma vez que cada máquina
possui, além dos cálculos sendo realizados, seus processos internos de
gerenciamento do sistema operacional, o que provavelmente ira dividir os
ciclos de processamento com alguns destes cálculos de docking simultâneos.

65
Tabela 7 - Tempos de execução individual de cada cálculo de docking, no AutoDock, ao utilizar
uma máquina e uma instância do programa AutoDock por máquina. As colunas E1 a E10
representa o tempo de execução individual de cada cópia de cada complexo dentro do mesmo
job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1abe 192 197 194 196 195 192 194 195 193 193 194.1 1.66 1acj 160 161 157 157 156 154 155 156 157 158 157.1 2.13
1acm 269 285 271 271 272 274 281 284 282 283 277.2 6.32 1aha 182 182 182 182 182 180 183 187 185 185 183.0 2.05 1baf 567 596 567 573 566 568 596 589 596 596 581.4 14,19 1cbs 332 333 323 319 324 319 334 333 333 334 328.4 6,36 1com 249 250 248 250 243 245 250 253 252 251 249.1 3,07 1dbb 209 226 218 216 208 209 224 226 224 226 218.6 7,65 1eap 410 428 410 417 417 412 430 430 428 428 421.0 8,59 1eed 212 218 212 214 214 212 215 217 216 214 214.4 2,12 1fki 198 211 198 199 196 198 209 208 210 209 203.6 6,20 1hri 358 370 360 364 357 367 372 373 373 372 366.6 6,40 1hsl 218 217 218 217 217 219 224 218 218 217 218.3 2,11 1igj 609 641 614 611 613 613 641 640 640 640 626,2 15,03 1ive 259 263 257 257 259 258 263 264 264 264 260,8 3,05 1lah 231 239 231 235 231 236 238 248 238 234 236,1 5,17 1lst 252 260 253 251 261 251 263 259 258 260 256,8 4,56
1mcr 141 146 143 140 141 151 145 146 144 147 144,4 3,34 1mrg 182 184 186 181 182 183 185 184 184 185 183,6 1,58 1mrk 385 401 384 400 387 384 404 404 401 403 395,3 8,99 1mup 155 156 157 156 156 157 157 159 154 158 156,5 1,43 1rob 396 415 400 401 403 398 412 414 413 419 407,1 8,30 1snc 402 424 405 405 402 406 424 426 427 424 414,5 11,18 1srj 364 377 363 363 374 363 378 377 380 377 371,6 7,34 1stp 270 285 276 276 270 272 284 283 285 284 278,5 6,36 1tdb 319 331 326 318 324 320 331 332 333 332 326,6 5,97 1tng 170 173 178 171 173 171 170 171 171 170 171,8 2,44 1tnl 189 188 183 182 183 184 187 188 187 187 185,8 2,53 1tph 189 194 190 192 188 189 198 196 193 193 192,2 3,26 1tpp 306 321 313 304 306 306 322 323 321 320 314,2 7,97
1ukz 607 631 631 595 596 600 631 629 630 632 618,2 16,42 1ulb 154 159 153 164 156 155 160 161 160 160 158,2 3,52
2ack 184 186 183 184 184 184 184 184 184 185 184,2 0,79
2ak3 442 466 443 446 444 456 464 463 464 464 455,2 10,24
2cgr 510 529 519 511 517 507 533 527 530 531 521,4 9,78 2cht 212 217 214 212 213 212 219 215 218 214 214,6 2,59 2cmd 240 242 246 244 239 241 244 244 244 243 242,7 2,16 2dbl 367 386 376 366 368 368 383 383 384 383 376,4 8,29
2gbp 234 234 241 235 234 232 235 235 237 233 235,0 2,49 2r07 435 453 460 434 436 453 453 455 453 454 448,6 9,63
2sim 394 414 393 395 392 391 409 413 409 409 401,9 9,58 3hvt 211 225 218 213 210 214 228 227 225 223 219,4 6,98
3ptb 1808 1865 1808 1797 1820 1812 1847 1831 1861 1850 1829,9 24,40
3tpi 179 179 176 179 176 179 180 179 178 178 178,3 1,34 4cts 166 168 166 166 165 167 167 166 167 168 166,6 0,97 4fab 337 351 338 351 337 352 350 352 352 352 347,2 6,84 6abp 133 137 136 137 134 134 135 134 134 134 134,8 1,40 7tim 198 206 197 203 205 210 207 206 208 206 204,6 4,17 8atc 273 284 273 273 272 283 282 283 283 284 279,0 5,42 8gch 123 120 120 123 120 121 122 122 121 122 121,4 1,17

66
. Tabela 8 - Tempos de execução individual de cada cálculo de docking, no AutoDock, ao
utilizar uma máquina e oito instâncias simultâneas do programa AutoDock por máquina. As
colunas E1 a E10 representa o tempo de execução individual de cada cópia de cada complexo
dentro do mesmo job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1abe 204 189 205 210 207 202 207 203 204 198 202,9 5,86 1acj 178 158 173 176 168 169 174 167 167 170 170,0 5,70
1acm 280 282 293 285 281 276 284 286 282 284 283,3 4,45 1aha 201 187 188 183 188 181 181 186 187 180 186,2 6,05
1baf 616 606 615 611 616 614 608 611 620 610 612,7 4,24 1cbs 340 336 339 337 332 332 339 338 330 336 335,9 3,45 1com 262 256 259 254 253 251 260 260 257 254 256,6 3,60 1dbb 243 231 236 239 227 236 235 237 230 235 234,9 4,60 1eap 442 440 448 442 442 449 448 443 441 442 443,7 3,30
1eed 233 226 231 229 227 225 236 224 226 228 228,5 3,81 1fki 232 230 238 230 228 237 227 234 235 230 232,1 3,75 1hri 397 390 390 390 388 384 387 388 380 383 387,7 4,69
1hsl 257 256 258 261 255 252 256 258 258 253 256,4 2,63 1igj 640 626 631 632 632 628 631 645 634 629 632,8 5,71 1ive 289 277 287 282 284 281 282 284 279 284 282,9 3,54 1lah 258 254 260 258 262 255 252 259 255 261 257,4 3,27
1lst 278 283 279 283 281 276 280 279 284 280 280,3 2,50 1mcr 158 155 160 157 156 157 158 158 158 157 157,4 1,35 1mrg 196 193 197 193 194 192 192 194 193 197 194,1 1,91 1mrk 430 430 430 436 435 434 432 429 422 424 430,2 4,49 1mup 174 168 169 167 170 171 169 168 169 174 169,9 2,42 1rob 439 425 435 440 430 432 435 427 434 426 432,3 5,25 1snc 463 446 454 461 461 454 459 450 461 451 456,0 5,79 1srj 391 392 394 396 393 385 378 389 374 386 387,8 7,15 1stp 313 314 311 312 300 307 312 311 319 308 310,7 4,99 1tdb 344 339 342 344 353 343 336 344 347 333 342,5 5,60 1tng 182 186 181 190 180 182 189 185 185 185 184,5 3,31 1tnl 198 197 193 196 200 199 196 195 207 197 197,8 3,79 1tph 213 206 203 207 206 203 211 201 205 203 205,8 3,76 1tpp 322 329 331 322 325 325 325 318 318 331 324,6 4,74 1ukz 586 586 593 585 594 585 587 586 588 594 588,4 3,75 1ulb 173 177 179 174 173 174 175 176 177 171 174,9 2,38 2ack 197 194 194 194 192 200 197 196 198 197 195,9 2,38 2ak3 493 483 492 495 488 488 486 494 483 484 488,6 4,62 2cgr 550 548 559 550 569 560 559 556 552 556 555,9 6,28 2cht 235 234 235 236 233 238 237 240 237 236 236,1 2,02 2cmd 250 251 248 250 253 252 256 251 253 251 251,5 2,17 2dbl 414 408 407 416 413 408 405 402 408 407 408,8 4,29 2gbp 265 258 257 261 262 261 258 251 256 258 258,7 3,83 2r07 483 477 491 486 486 477 480 475 478 476 480,9 5,34 2sim 418 418 427 418 418 417 417 417 417 422 418,9 3,21 3hvt 231 233 236 230 232 232 238 231 235 238 233,6 2,95 3ptb 1828 1807 1837 1801 1805 1839 1816 1802 1796 1796 1812,7 16,42 3tpi 186 187 189 191 190 186 180 191 186 187 187,3 3,27 4cts 171 172 166 170 169 171 166 168 168 166 168,7 2,26 4fab 362 359 367 354 360 357 361 353 362 357 359,2 4,16 6abp 148 150 147 153 150 145 146 149 155 148 149,1 3,07 7tim 210 213 206 204 209 209 212 210 204 213 209,0 3,37 8atc 287 258 282 282 276 283 287 274 280 284 279,3 8,58 8gch 138 121 138 133 132 133 130 133 130 134 132,2 4,80

67
Tabela 9 - Tempos de execução individual de cada cálculo de docking, no AutoDock, ao utilizar
seis máquinas e apenas uma instância do programa AutoDock por máquina. As colunas E1 a
E10 representa o tempo de execução individual de cada cópia de cada complexo dentro do
mesmo job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1abe 193 192 193 193 193 193 193 193 191 193 192,7 0,67 1acj 158 155 154 157 155 156 157 155 156 157 156,0 1,25
1acm 277 279 277 279 278 279 284 278 283 279 279,3 2,36 1aha 184 183 184 184 184 185 181 183 184 184 183,6 1,07 1baf 585 592 593 591 583 589 599 598 594 587 591,1 5,24 1cbs 329 330 329 333 329 332 329 334 329 330 330,4 1,90 1com 247 251 248 252 251 253 251 250 251 252 250,6 1,84 1dbb 223 214 224 221 220 223 223 221 227 229 222,5 4,06 1eap 422 423 427 426 429 429 428 427 431 422 426,4 3,13 1eed 214 214 214 214 215 214 214 212 214 213 213,8 0,79 1fki 206 208 207 209 210 205 204 208 204 202 206,3 2,54 1hri 372 354 373 368 368 374 371 368 371 370 368,9 5,65 1hsl 231 230 231 230 228 231 237 230 231 230 230,9 2,33 1igj 627 637 631 641 644 630 629 642 628 629 633,8 6,51 1ive 261 259 261 261 259 263 264 261 264 262 261,5 1,78 1lah 233 234 236 237 234 234 237 235 236 235 235,1 1,37 1lst 257 255 261 255 255 260 257 254 256 258 256,8 2,30
1mcr 142 141 145 144 144 145 144 145 146 145 144,1 1,52 1mrg 185 182 184 181 182 184 182 183 185 190 183,8 2,57 1mrk 396 388 396 400 403 395 397 399 396 404 397,4 4,53 1mup 158 156 158 155 159 157 156 155 155 155 156,4 1,50 1rob 409 410 411 407 407 413 407 407 408 407 408,6 2,12 1snc 422 419 415 418 423 419 424 418 417 420 419,5 2,80 1srj 379 366 372 373 377 370 372 380 372 380 374,1 4,70 1stp 281 282 287 281 284 287 282 281 287 281 283,3 2,71 1tdb 326 326 325 329 327 330 332 328 326 327 327,6 2,17 1tng 184 181 179 183 183 180 183 182 182 180 181,7 1,64 1tnl 188 184 187 185 186 186 185 188 185 188 186,2 1,47 1tph 192 192 190 194 197 192 191 192 192 192 192,4 1,90 1tpp 321 322 324 315 321 317 321 319 319 318 319,7 2,63 1ukz 615 617 618 618 615 619 619 620 617 626 618,4 3,13 1ulb 157 157 157 156 156 157 159 158 157 160 157,4 1,26 2ack 183 185 185 183 183 185 187 183 184 183 184,1 1,37 2ak3 463 456 465 467 462 457 461 457 459 462 460,9 3,63 2cgr 523 528 533 532 522 530 527 529 531 522 527,7 4,11 2cht 216 216 213 213 215 217 213 214 213 214 214,4 1,50 2cmd 240 242 246 243 241 244 243 242 244 241 242,6 1,78 2dbl 380 372 376 378 377 378 382 377 376 382 377,8 3,01 2gbp 234 233 234 236 234 232 235 237 236 237 234,8 1,69 2r07 446 452 448 449 449 456 447 451 453 449 450,0 3,02 2sim 406 403 410 409 412 403 407 407 408 409 407,4 2,87 3hvt 223 208 220 221 222 220 221 220 220 225 220,0 4,52 3ptb 1819 1816 1814 1857 1875 1862 1856 1844 1845 1835 1842,3 20,98 3tpi 180 180 178 179 177 180 178 177 180 178 178,7 1,25 4cts 166 167 166 166 166 167 167 165 167 168 166,5 0,85 4fab 344 345 351 349 352 345 360 349 351 350 349,6 4,62 6abp 134 134 134 134 136 134 132 134 135 132 133,9 1,20 7tim 208 205 204 203 205 205 206 204 206 205 205,1 1,37 8atc 278 283 278 278 280 282 281 284 279 280 280,3 2,16 8gch 121 124 121 120 120 120 122 124 121 122 121,5 1,51

68
Figura 5.7 – Gráfico de dispersão dos cálculos individuais de docking com o programa
AutoDock ao utilizar: (a) uma máquina e uma instância de execução; (b) uma máquina e oito
instâncias de execução; (c) seis máquinas e uma instância de execução.

69
As pequenas barras mostradas nos gráficos da Figura 5.7 representam o
desvio padrão ao redor da média de tempo das 10 cópias de execução do
cálculo de docking para cada complexo. Conforme se pode observar nestes
gráficos, o desvio padrão observado para estas amostras de tempo é baixo, em
relação ao tempo total de execução de cada cálculo individual. Isto indica que
estes cálculos de docking realizados nestas configurações e ajustes (“setup”)
do programa AutoDock, parecem ser significativamente reprodutíveis,
particularmente no que diz respeito à demanda computacional que eles
apresentam. Ao se levar em consideração os cálculos com seis máquinas
(Tabela 9) (Figura 5.7c), também são observados valores baixos de desvio
padrão. Neste cálculo de docking com seis máquinas há uma grande chance
de que as cópias de execução de cada cálculo tenham sido realizadas por
máquinas diferentes, reforçando ainda mais a reprodutibilidade dos cálculos
individuais, permitindo que a comparação de desempenho seja fundamentada
em critérios quantitativos.
Acredita-se que estes tempos de execução podem ter sido obtidos
devido a mudança realizada no parâmetro seed (semente inicial para o gerador
de números aleatórios) para um valor fixo. Este parâmetro determina por onde
o algoritmo genético começará sua busca e, ao torná-lo fixo, possibilita a
reprodutibilidade dos procedimentos de cálculo de docking, uma vez que o
cálculo sempre começará da mesma maneira. Desta forma acredita-se ser
possível a reprodução destes tempos, desde que nas mesmas circunstâncias
de configuração, quantidade de máquinas e instâncias de execução do
programa AutoDock.
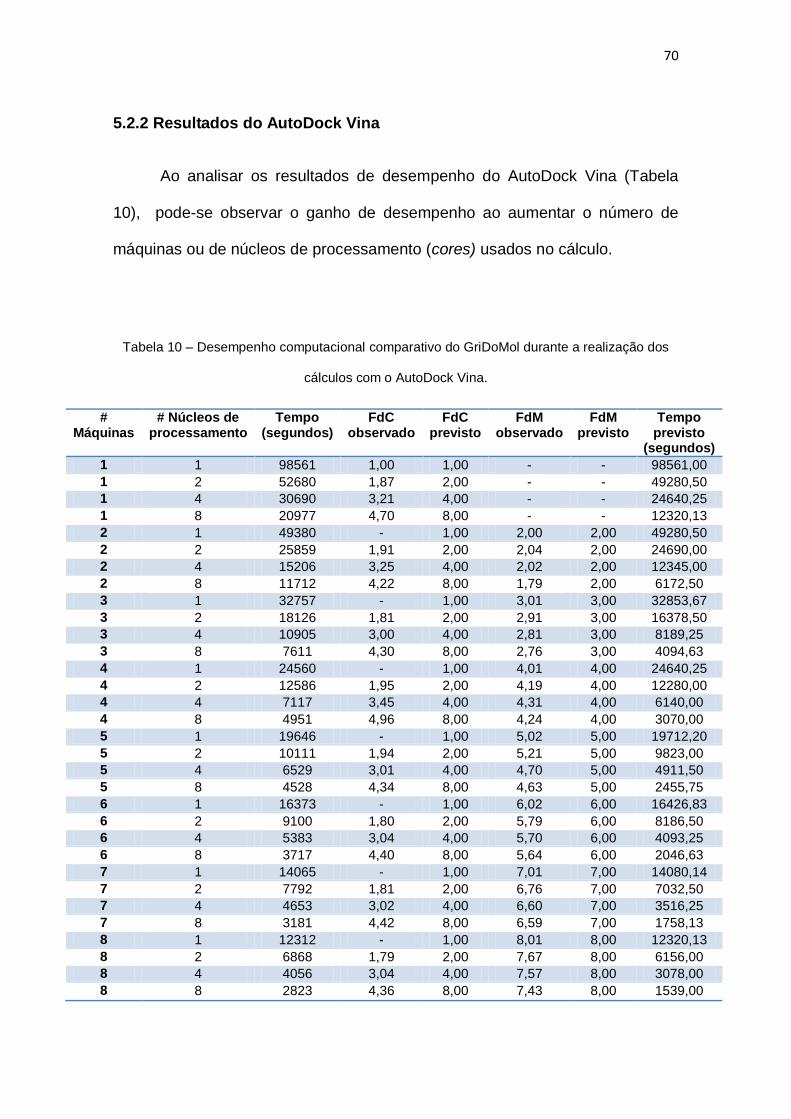
70
5.2.2 Resultados do AutoDock Vina
Ao analisar os resultados de desempenho do AutoDock Vina (Tabela
10), pode-se observar o ganho de desempenho ao aumentar o número de
máquinas ou de núcleos de processamento (cores) usados no cálculo.
Tabela 10 – Desempenho computacional comparativo do GriDoMol durante a realização dos
cálculos com o AutoDock Vina.
# Máquinas
# Núcleos de processamento
Tempo (segundos)
FdC observado
FdC previsto
FdM observado
FdM previsto
Tempo previsto
(segundos)
1 1 98561 1,00 1,00 - - 98561,00
1 2 52680 1,87 2,00 - - 49280,50
1 4 30690 3,21 4,00 - - 24640,25
1 8 20977 4,70 8,00 - - 12320,13
2 1 49380 - 1,00 2,00 2,00 49280,50
2 2 25859 1,91 2,00 2,04 2,00 24690,00
2 4 15206 3,25 4,00 2,02 2,00 12345,00
2 8 11712 4,22 8,00 1,79 2,00 6172,50
3 1 32757 - 1,00 3,01 3,00 32853,67
3 2 18126 1,81 2,00 2,91 3,00 16378,50
3 4 10905 3,00 4,00 2,81 3,00 8189,25
3 8 7611 4,30 8,00 2,76 3,00 4094,63
4 1 24560 - 1,00 4,01 4,00 24640,25
4 2 12586 1,95 2,00 4,19 4,00 12280,00
4 4 7117 3,45 4,00 4,31 4,00 6140,00
4 8 4951 4,96 8,00 4,24 4,00 3070,00
5 1 19646 - 1,00 5,02 5,00 19712,20
5 2 10111 1,94 2,00 5,21 5,00 9823,00
5 4 6529 3,01 4,00 4,70 5,00 4911,50
5 8 4528 4,34 8,00 4,63 5,00 2455,75
6 1 16373 - 1,00 6,02 6,00 16426,83
6 2 9100 1,80 2,00 5,79 6,00 8186,50
6 4 5383 3,04 4,00 5,70 6,00 4093,25
6 8 3717 4,40 8,00 5,64 6,00 2046,63
7 1 14065 - 1,00 7,01 7,00 14080,14
7 2 7792 1,81 2,00 6,76 7,00 7032,50
7 4 4653 3,02 4,00 6,60 7,00 3516,25
7 8 3181 4,42 8,00 6,59 7,00 1758,13
8 1 12312 - 1,00 8,01 8,00 12320,13
8 2 6868 1,79 2,00 7,67 8,00 6156,00
8 4 4056 3,04 4,00 7,57 8,00 3078,00
8 8 2823 4,36 8,00 7,43 8,00 1539,00
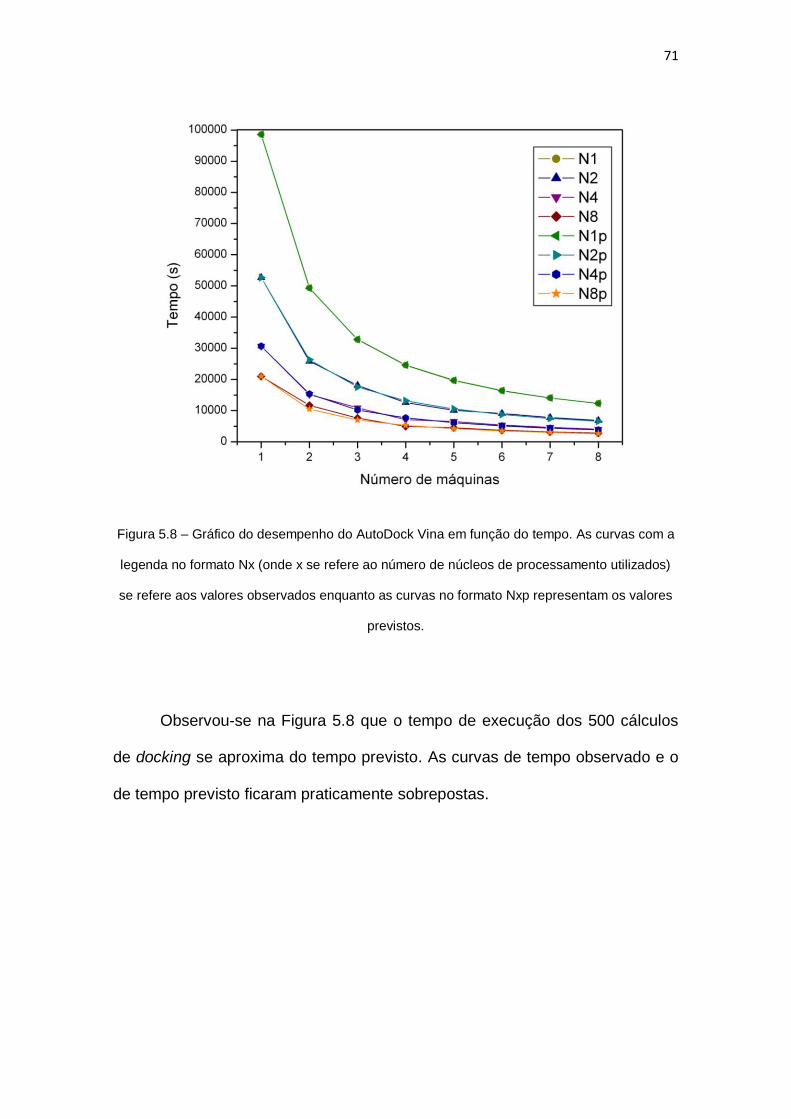
71
Figura 5.8 – Gráfico do desempenho do AutoDock Vina em função do tempo. As curvas com a
legenda no formato Nx (onde x se refere ao número de núcleos de processamento utilizados)
se refere aos valores observados enquanto as curvas no formato Nxp representam os valores
previstos.
Observou-se na Figura 5.8 que o tempo de execução dos 500 cálculos
de docking se aproxima do tempo previsto. As curvas de tempo observado e o
de tempo previsto ficaram praticamente sobrepostas.

72
Figura 5.9 – Gráfico dos valores de FdC no programa AutoDock Vina. A legenda no formato Mx
(onde x se refere ao número de máquinas utilizadas) se refere aos valores observados.
Pode-se observar no gráfico do FdC obtido para o programa AutoDock
Vina (Figura 5.9), o comportamento mais homogêneo dos valores previstos e
observados de tempo de cálculo total para cada teste. Verificou-se, nesta
métrica, valores aquém do valor teórico esperado ou previsto para FdC, ficando
mais evidente nos casos que utilizam oito núcleos de processamento, os quais,
nestes casos, atingem valores um pouco mais da metade do previsto para FdC.
Isto ocorre, devido ao fato de que o aumento do número de núcleos de
processamento reduz o tempo sequencial com processamento, mas
tipicamente não reduz o tempo gasto com as outras operações fundamentais

73
que precisam ser realizadas no ambiente de grid computacional. Desta forma,
o número de núcleos de processamento (cores) só acelera a execução do
cálculo de docking na máquina, não influenciando a parcela de tempo gasto
pela comunicação e transferências de arquivos entre os componentes do grid
computacional. Além disto, acredita-se que esta diferença entre o desempenho
previsto e observado esteja relacionada a limitação de ganho de desempenho
na paralelização interna do programa AutoDock Vina. Uma discussão mais
detalhada e quantitativa deste efeito encontra-se no final da seção 5.2.2.1.
Figura 5.10 – Gráfico dos valores de FdM no programa AutoDock Vina. A legenda no formato
Mx (onde x se refere ao número de máquinas utilizadas) se refere aos valores observados.
Os valores do FdM para o programa AutoDock Vina (Figura 5.10), em
geral, se mantiveram próximos aos valores previstos. Entretanto, alguns

74
valores para o FdM ficaram acima do previsto, como, por exemplo, ao utilizar
cinco máquinas e dois dos núcleos de processamento em cada máquina (barra
N2, com cinco máquinas). Outro exemplo de teste onde este efeito pode ser
observado foi com quatro máquinas e quatro núcleos de processamento cada
(barra N4 com quatro máquinas). Isto ocorre devido a ocasionais transferências
simultâneas, as quais reduziram o tempo total gasto na transferência de
arquivos e, consequentemente, diminuiu o tempo total de execução do job
(conjunto de cálculos) como um todo.
Para elucidar esta hipótese sobre a transferência simultânea impactar o
FdM, selecionou-se alguns destes testes que apresentaram valores de FdM
observado acima do previsto, barras N2 com cinco máquinas e N4 com quatro
máquinas, foram refeitos sem a etapa de transferência de arquivos de tal forma
que todos os arquivos necessários para a realização dos cálculos foram pré-
disponibilizados em cada máquina e, desta forma, obteve-se uma redução dos
valores de FdM observados para valores mais próximos do previsto (Tabela
11).
Tabela 11 – Cálculos refeitos para elucidar os valores elevados de FdM encontrados em alguns
dos cálculos.
Máquinas (X)
Núcleos de processamento
FdM com transferência
(TXct)
Tempo com transferência
(s)
FdM sem transferência
(TXst)
Tempo sem transferência
(s)
Diferença percentual
1 2 - 52680 - 49585 5,87%
1 4 - 30690 - 27809 9,39%
4 4 4,31 7117 4,10 6783 4,69%
5 2 5,21 10111 5,07 9784 3,24%

75
A diferença percentual entre os cálculos com a transferência de arquivos
e os cálculos com todos os arquivos pré-disponibilizados em cada máquina,
eliminando-se desta forma, a transferência principal de arquivos, dá uma idéia
da percentagem de tempo gasto com esta etapa de transferências (Tabela 11).
Esta comparação percentual relativa é calculada através da diferença entre o
tempo de execução com a etapa de transferência de arquivos (TXct) e o tempo
de execução sem esta etapa (TXst), dividido pelo tempo de execução com
transferência de arquivos (TXct), conforme observa-se na equação 3.
Diferença percentual do cálculo com X máquinas = (TXct – TXst)/TXct (3)
Esta diferença percentual relativa indica o quanto a execução do
conjunto de cálculos demorou em relação a seu análogo sem a etapa de
transferência de arquivos. Por exemplo, o cálculo com a etapa de transferência
de arquivos realizado em uma máquina utilizando quatro dos oito núcleos de
processamento é 9,39% maior do que o seu análogo sem a transferência de
arquivos. Esta diferença diminui ao aumentar o número de máquinas utilizadas
na execução do job (conjunto de cálculos), como é o caso de quatro e cinco
máquinas, com diferenças percentuais de 4,69 e 3,24%, respectivamente.
Acredita-se que isto pode ocorrer devido à redução do tempo gasto com as
comunicações e transferências de arquivos quando estas ocorrem de forma
simultânea, em ambientes onde várias máquinas estão sendo utilizadas na
distribuição das tarefas.
Em adição, os dados da Tabela 11 também evidenciam que os valores
de FdM observados se aproximaram dos valores previstos, quando a etapa de

76
transferência de arquivos foi suprimida, ou seja, quando os arquivos eram pré-
disponibilizados nas máquinas de cálculo. Em outras palavras, o valor de FdM
observado com transferência de arquivos (cálculo padrão) para o caso de
quatro máquinas e quatro núcleos de processamento, foi de 4,31. Ao se
eliminar a etapa de transferência de arquivos, o valor de FdM observado foi
para o valor de 4,10 mais próximo ao valor previsto, igual a 4. O mesmo
acontece com o caso de 5 máquinas e 2 cores, o valor de FdM observado com
transferência de arquivos é de 5,21, enquanto o valor observado sem
transferência de arquivos é de 5,07, mais próximo do valor previsto, igual a 5.
Estas observações nos valores observados de FdM e como eles diminuem, se
aproximando dos valores previstos, quando se realiza o procedimento sem a
transferência de arquivos, corrobora com a hipótese levantada e exposta nos
parágrafos acima.
Examinando-se todos os testes do GriDoMol com o programa AutoDock
Vina e considerando o desempenho do ambiente de grid computacional como
um todo, pode-se observar que ao utilizar mais máquinas, ao invés de mais
núcleos de processamento, o processo mostrou-se mais eficiente, levando
menor quantidade total de tempo. Por exemplo, utilizar oito máquinas e apenas
um núcleo de processamento (ver Tabela 10 - tempo de execução: 12.312
segundos) é mais rápido do que utilizar uma máquina com oito núcleos de
processamento (cores) (ver Tabela 10 - tempo de execução: 20.977 segundos).
Ao se utilizar ambas as formas de distribuição para a realização dos
cálculos de docking, o ambiente de grid computacional e a paralelização
multicore, pode ser obtida uma redução do tempo total necessário para apenas

77
3% do tempo necessário comparado com o cálculo não paralelizado. Desta
forma, a utilização destas formas de distribuição proporcionou um aumento de
desempenhos de até 34,91 vezes, com cálculos mais rápidos no ambiente de
grid computacional acessado pelo GriDoMol.
5.2.2.1 Reprodutibilidade do tempo de execução do AutoDock Vina
Com o intuito de determinar se os parâmetros utilizados nos cálculos de
docking com o programa AutoDock Vina tornou os cálculos individuais
reprodutíveis em demanda computacional, foi analisado o tempo individual de
execução de cada um destes cálculos para uma máquina utilizando apenas um
núcleo de processamento (Tabela 12), uma máquina utilizando oito núcleos de
processamento (Tabela 13) e oito máquinas utilizando apenas um núcleo de
processamento (Tabela 14). Esta escolha de número de máquinas e núcleos
de processamento utilizados foi feita com o intuito de analisar possíveis
impactos no tempo de execução individual de cada cálculo ao aumentar o
número de máquinas ou núcleos de processamento. Os dados de tempo
utilizados nesta analise foram obtidos através dos arquivos de saída dos
cálculos individuais.
Observa-se nestas tabelas (Tabela 12, 13 e 14) que os valores obtidos
em cada cópia do cálculo se aproxima da média e que o desvio padrão nestes
casos é baixo, levando em consideração o tempo individual de execução
destes cálculos.

78
Tabela 12 – Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar uma máquina e um núcleo de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1abe 70,07 69,97 70,03 70,02 70,01 70,11 70,02 70,10 70,04 70,06 70,04 0,04 1acj 38,82 39,43 38,83 38,85 38,85 39,44 38,87 39,43 38,84 38,84 39,02 0,28
1acm 153,50 153,47 154,37 153,54 153,50 154,74 153,58 153,56 153,52 153,52 153,73 0,44 1aha 41,34 40,95 41,34 41,34 41,35 41,36 40,99 40,98 40,97 40,96 41,16 0,20 1baf 546,50 546,44 549,53 549,55 546,70 549,51 546,84 546,83 546,35 547,10 547,54 1,39 1cbs 171,48 171,47 172,83 171,44 171,47 171,45 171,47 172,79 172,75 171,49 171,86 0,64 1com 165,58 165,49 166,76 165,55 165,58 165,63 165,70 166,79 165,86 165,59 165,85 0,50 1dbb 162,56 162,59 165,64 162,66 162,61 162,60 162,63 165,59 162,57 162,66 163,21 1,27 1eap 359,23 360,74 359,31 359,35 359,16 359,20 360,84 359,36 359,31 359,42 359,59 0,64 1eed 134,72 133,92 133,88 134,75 133,92 133,86 134,71 133,92 133,90 134,50 134,21 0,40 1fki 92,67 90,87 90,86 90,88 90,94 92,70 90,93 90,89 90,86 90,85 91,25 0,76 1hri 461,42 461,54 461,81 461,33 461,79 461,62 461,57 463,60 463,44 461,56 461,97 0,83 1hsl 93,00 92,93 93,50 92,94 92,90 93,46 92,96 93,02 92,95 93,49 93,12 0,26 1igj 620,65 616,67 616,66 616,88 616,55 616,93 616,57 616,64 620,87 616,69 617,51 1,72 1ive 239,48 240,77 239,51 239,39 239,58 239,52 239,46 239,50 239,50 239,43 239,61 0,41 1lah 99,43 99,02 99,03 99,01 98,96 99,46 99,06 99,00 99,45 98,97 99,14 0,21 1lst 125,95 126,47 125,94 126,44 125,93 126,46 125,96 125,93 125,93 125,89 126,09 0,25
1mcr 32,58 32,60 32,60 32,97 32,97 32,60 32,61 32,59 32,60 32,60 32,67 0,16 1mrg 49,72 49,72 49,69 49,72 50,04 49,68 49,72 49,73 49,71 50,25 49,80 0,19 1mrk 233,31 234,63 233,30 233,36 233,31 233,40 234,72 233,34 233,36 233,40 233,61 0,56 1mup 33,61 33,36 33,31 33,33 33,34 33,60 33,35 33,30 33,36 33,30 33,39 0,12 1rob 256,81 256,78 256,72 256,71 258,95 256,76 256,76 256,78 258,31 256,79 257,14 0,80 1snc 362,41 360,15 360,32 360,29 360,21 362,40 360,33 360,51 360,41 360,08 360,71 0,90 1srj 167,84 169,09 167,93 167,85 167,88 167,87 167,82 167,93 169,03 167,86 168,11 0,50 1stp 154,97 154,95 153,82 172,21 153,85 154,92 153,90 154,95 153,86 153,89 156,13 5,67 1tdb 183,00 182,96 183,04 183,09 183,12 184,34 183,38 184,31 183,21 183,03 183,35 0,53 1tng 37,30 41,95 37,30 37,33 37,32 37,31 37,35 37,31 37,72 37,32 37,82 1,46 1tnl 45,88 45,87 45,49 45,39 45,85 45,42 46,00 45,48 45,56 45,42 45,64 0,23 1tph 69,41 69,45 69,48 70,07 69,48 69,45 70,02 69,49 69,46 69,40 69,57 0,25
1tpp 173,39 173,42 175,32 173,56 173,38 173,36 175,47 173,46 174,60 173,38 173,93 0,85 1ukz 534,82 535,56 535,29 535,22 535,61 537,66 535,10 535,45 535,27 535,49 535,55 0,78 1ulb 32,62 32,30 32,62 32,61 32,21 32,21 32,22 32,24 32,58 32,23 32,38 0,19 2ack 56,13 56,08 56,13 56,13 56,08 56,78 56,17 56,74 56,12 56,08 56,24 0,27 2ak3 321,01 320,87 320,69 321,00 320,86 320,78 320,91 321,27 320,75 321,90 321,00 0,35 2cgr 399,73 399,75 397,42 397,44 397,71 397,53 399,79 397,44 397,44 397,52 398,18 1,09 2cht 80,39 79,64 79,67 80,42 79,72 80,42 80,45 80,42 80,42 80,46 80,20 0,36 2cmd 122,68 123,39 122,66 122,64 122,66 122,69 123,28 122,69 122,65 122,68 122,80 0,28 2dbl 320,80 320,93 320,93 320,86 320,94 320,89 323,38 320,89 320,86 320,85 321,13 0,79 2gbp 167,02 166,85 166,86 166,83 168,14 166,88 166,88 168,14 166,94 166,80 167,13 0,53 2r07 399,57 399,73 400,24 401,82 400,05 400,02 400,05 399,69 401,75 402,06 400,50 0,97 2sim 277,63 277,59 276,24 275,91 275,91 275,96 276,12 276,09 275,97 276,05 276,35 0,67 3hvt 73,25 73,23 73,25 73,55 73,28 73,31 74,18 73,27 74,15 73,25 73,47 0,37 3ptb 40,42 40,47 40,42 40,43 40,87 40,86 40,87 40,87 40,42 40,41 40,60 0,23 3tpi 55,37 55,39 55,43 55,75 55,37 55,39 55,42 55,74 55,38 55,40 55,46 0,15 4cts 44,44 44,81 44,87 44,45 44,45 44,48 44,43 44,98 44,45 44,45 44,58 0,21 4fab 192,11 192,24 191,04 190,95 192,26 190,84 192,43 193,22 192,40 192,06 191,96 0,77 6abp 20,31 20,30 20,39 20,32 20,30 20,29 20,60 20,31 20,60 20,79 20,42 0,18 7tim 74,35 74,44 74,36 74,38 74,40 74,39 74,47 74,38 74,38 74,37 74,39 0,04 8atc 143,64 143,64 143,61 144,51 143,64 143,57 143,68 144,45 143,67 143,65 143,81 0,36 8gch 14,42 14,45 14,42 14,32 14,43 14,29 14,34 14,43 14,30 14,43 14,38 0,06

79
Tabela 13 – Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar uma máquina e oito núcleos de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1ABE 11,43 15,39 12,07 15,23 10,37 15,43 10,03 15,21 10,57 14,55 13,03 2,33 1ACJ 6,18 5,91 7,48 6,02 5,95 6,17 6,68 6,32 6,34 6,11 6,32 0,47 1ACM 22,31 22,29 22,12 22,64 22,21 22,47 22,33 22,12 22,29 22,12 22,29 0,17 1AHA 6,64 6,69 5,85 7,01 6,42 8,71 7,09 6,73 5,76 8,34 6,92 0,95 1BAF 73,44 74,89 74,89 75,17 74,80 74,82 74,70 74,17 75,15 74,65 74,67 0,51 1CBS 25,21 25,25 25,14 25,15 25,92 25,56 25,42 25,36 25,22 25,91 25,41 0,29 1COM 25,01 24,28 27,66 24,57 24,37 24,33 23,60 24,73 24,34 24,49 24,74 1,09 1DBB 22,64 22,29 22,54 22,58 22,59 22,45 22,56 22,61 22,65 22,09 22,50 0,18 1EAP 48,60 49,65 50,85 49,11 49,92 48,93 50,46 49,36 50,70 49,25 49,68 0,77 1EED 19,48 19,89 19,53 19,63 19,55 19,32 19,53 19,28 19,35 19,87 19,54 0,21 1FKI 15,69 16,00 16,03 15,64 20,30 16,64 16,10 15,98 16,31 16,25 16,49 1,37 1HRI 63,48 63,62 63,86 63,56 63,60 63,58 63,83 63,67 63,47 63,84 63,65 0,14 1HSL 15,54 12,90 15,45 15,51 15,56 15,72 15,57 15,53 15,59 15,63 15,30 0,85 1IGJ 84,17 83,94 83,95 84,44 84,02 84,15 84,08 84,01 84,07 84,78 84,16 0,26 1IVE 32,53 32,35 34,23 33,16 32,79 32,68 32,61 32,62 32,72 32,97 32,87 0,53 1LAH 17,13 17,90 14,98 17,94 18,08 13,73 16,30 13,34 16,25 17,94 16,36 1,79 1LST 20,04 19,96 20,25 19,91 20,08 19,62 19,98 20,01 20,04 19,98 19,99 0,16 1MCR 6,92 5,29 5,35 8,38 5,81 4,95 5,36 5,97 8,44 5,65 6,21 1,27 1MRG 9,11 10,67 9,66 11,20 11,10 7,38 9,92 7,12 9,74 7,46 9,34 1,54 1MRK 31,70 32,06 32,54 32,22 32,25 32,08 31,96 32,91 33,08 31,86 32,27 0,45 1MUP 5,49 4,92 6,04 5,21 5,11 6,07 5,37 5,19 6,29 5,17 5,49 0,47 1ROB 35,90 35,49 35,80 35,39 35,50 35,37 35,45 36,31 35,85 35,54 35,66 0,30 1SNC 49,15 49,49 49,15 49,24 49,19 49,15 49,47 49,13 48,83 49,14 49,19 0,18 1SRJ 26,43 25,96 25,87 26,09 26,00 25,81 25,87 26,65 27,15 26,18 26,20 0,43 1STP 22,68 21,27 22,68 23,73 22,88 22,57 22,63 22,61 22,54 22,41 22,60 0,59 1TDB 27,26 27,29 27,15 27,17 27,56 27,37 27,26 27,32 27,47 27,70 27,36 0,17 1TNG 6,16 7,29 6,09 5,88 7,64 6,47 6,08 7,64 7,33 7,77 6,84 0,76 1TNL 9,54 7,30 8,81 7,19 11,45 7,70 9,84 7,06 7,49 8,62 8,50 1,44 1TPH 12,96 11,57 14,66 11,81 15,00 13,52 14,44 14,73 11,96 13,70 13,44 1,30 1TPP 25,80 25,78 26,07 26,04 26,01 26,25 25,88 25,77 25,71 25,57 25,89 0,20 1UKZ 72,43 73,14 72,63 72,41 72,73 72,62 72,68 72,53 73,21 72,50 72,69 0,28 1ULB 6,24 5,97 6,15 5,80 7,65 6,27 5,97 6,44 6,05 6,00 6,25 0,52 2ACK 13,39 8,49 12,12 12,69 11,56 13,12 10,90 8,36 9,21 13,34 11,32 1,99 2AK3 46,06 45,48 45,22 45,68 44,36 46,24 45,62 45,73 46,14 46,07 45,66 0,56 2CGR 53,95 54,06 53,18 54,14 54,04 54,37 54,13 54,09 53,50 53,76 53,92 0,35 2CHT 11,27 14,66 14,67 11,54 11,71 12,93 14,76 14,63 14,68 14,24 13,51 1,48 2CMD 19,69 21,21 21,23 20,32 21,49 20,46 20,05 21,22 21,29 17,82 20,48 1,12 2DBL 43,16 44,13 44,57 44,28 43,03 44,59 44,33 44,59 43,16 45,42 44,13 0,78 2GBP 27,14 29,78 30,12 27,27 28,77 30,45 27,09 30,16 25,53 30,37 28,67 1,77 2R07 55,54 55,58 55,68 55,83 55,56 56,09 55,59 55,47 55,55 55,63 55,65 0,18 2SIM 39,56 39,22 39,19 38,99 38,87 39,21 38,63 39,59 39,07 38,78 39,11 0,31 3HVT 13,39 13,87 16,70 14,66 14,11 12,22 16,44 14,79 14,31 15,36 14,59 1,35 3PTB 9,65 6,94 7,24 9,04 7,97 8,29 9,75 7,89 6,75 6,55 8,01 1,17 3TPI 12,62 8,69 7,94 8,30 8,87 9,26 9,44 9,66 12,98 8,16 9,59 1,78 4CTS 6,50 8,85 7,13 8,53 8,42 9,04 7,74 6,88 10,36 6,75 8,02 1,23 4FAB 27,49 28,04 27,83 27,52 27,95 27,46 27,91 28,25 27,64 28,09 27,82 0,28 6ABP 3,78 3,97 3,63 3,85 3,51 3,73 3,74 3,56 3,99 3,84 3,76 0,16 7TIM 14,40 14,35 14,14 12,37 14,36 14,17 14,26 14,22 14,16 14,43 14,09 0,61 8ATC 22,58 22,30 22,42 22,21 22,36 22,41 22,27 22,27 22,43 22,33 22,36 0,11 8GCH 3,46 3,30 3,47 3,24 4,44 3,55 3,70 3,54 3,29 3,74 3,57 0,35
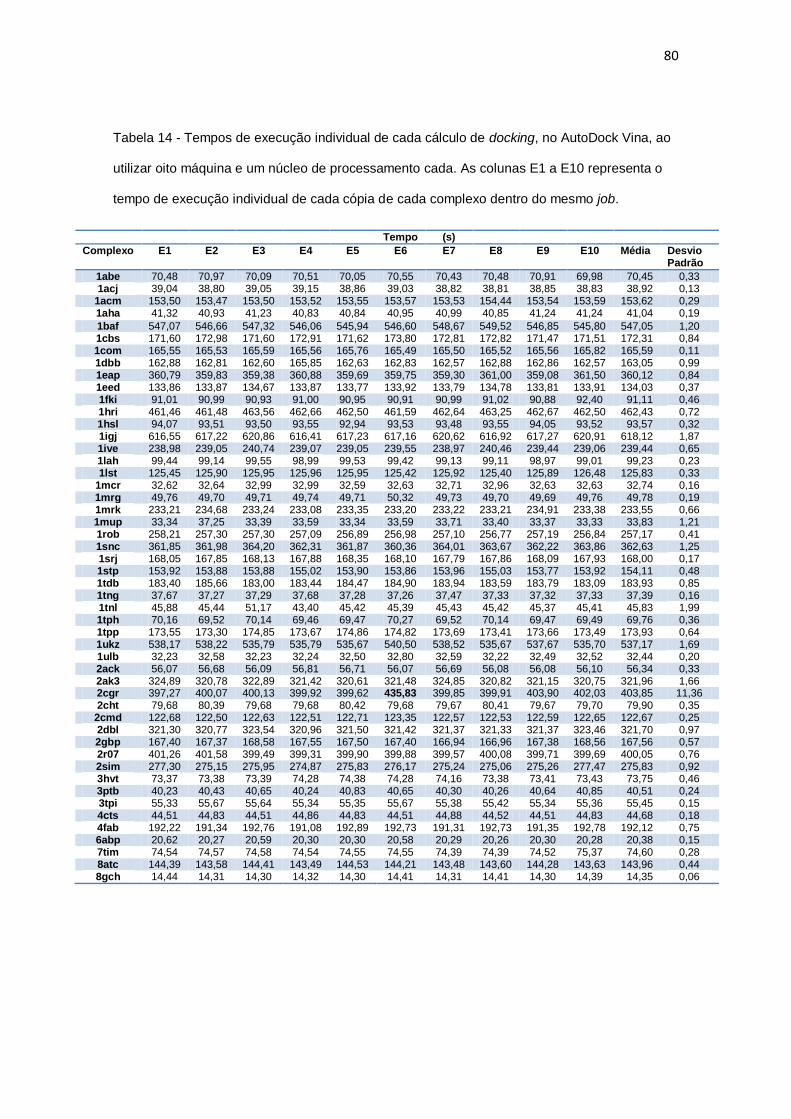
80
Tabela 14 - Tempos de execução individual de cada cálculo de docking, no AutoDock Vina, ao
utilizar oito máquina e um núcleo de processamento cada. As colunas E1 a E10 representa o
tempo de execução individual de cada cópia de cada complexo dentro do mesmo job.
Tempo (s)
Complexo E1 E2 E3 E4 E5 E6 E7 E8 E9 E10 Média Desvio Padrão
1abe 70,48 70,97 70,09 70,51 70,05 70,55 70,43 70,48 70,91 69,98 70,45 0,33 1acj 39,04 38,80 39,05 39,15 38,86 39,03 38,82 38,81 38,85 38,83 38,92 0,13
1acm 153,50 153,47 153,50 153,52 153,55 153,57 153,53 154,44 153,54 153,59 153,62 0,29 1aha 41,32 40,93 41,23 40,83 40,84 40,95 40,99 40,85 41,24 41,24 41,04 0,19
1baf 547,07 546,66 547,32 546,06 545,94 546,60 548,67 549,52 546,85 545,80 547,05 1,20 1cbs 171,60 172,98 171,60 172,91 171,62 173,80 172,81 172,82 171,47 171,51 172,31 0,84 1com 165,55 165,53 165,59 165,56 165,76 165,49 165,50 165,52 165,56 165,82 165,59 0,11 1dbb 162,88 162,81 162,60 165,85 162,63 162,83 162,57 162,88 162,86 162,57 163,05 0,99 1eap 360,79 359,83 359,38 360,88 359,69 359,75 359,30 361,00 359,08 361,50 360,12 0,84 1eed 133,86 133,87 134,67 133,87 133,77 133,92 133,79 134,78 133,81 133,91 134,03 0,37 1fki 91,01 90,99 90,93 91,00 90,95 90,91 90,99 91,02 90,88 92,40 91,11 0,46 1hri 461,46 461,48 463,56 462,66 462,50 461,59 462,64 463,25 462,67 462,50 462,43 0,72 1hsl 94,07 93,51 93,50 93,55 92,94 93,53 93,48 93,55 94,05 93,52 93,57 0,32 1igj 616,55 617,22 620,86 616,41 617,23 617,16 620,62 616,92 617,27 620,91 618,12 1,87 1ive 238,98 239,05 240,74 239,07 239,05 239,55 238,97 240,46 239,44 239,06 239,44 0,65 1lah 99,44 99,14 99,55 98,99 99,53 99,42 99,13 99,11 98,97 99,01 99,23 0,23 1lst 125,45 125,90 125,95 125,96 125,95 125,42 125,92 125,40 125,89 126,48 125,83 0,33
1mcr 32,62 32,64 32,99 32,99 32,59 32,63 32,71 32,96 32,63 32,63 32,74 0,16 1mrg 49,76 49,70 49,71 49,74 49,71 50,32 49,73 49,70 49,69 49,76 49,78 0,19 1mrk 233,21 234,68 233,24 233,08 233,35 233,20 233,22 233,21 234,91 233,38 233,55 0,66 1mup 33,34 37,25 33,39 33,59 33,34 33,59 33,71 33,40 33,37 33,33 33,83 1,21 1rob 258,21 257,30 257,30 257,09 256,89 256,98 257,10 256,77 257,19 256,84 257,17 0,41 1snc 361,85 361,98 364,20 362,31 361,87 360,36 364,01 363,67 362,22 363,86 362,63 1,25 1srj 168,05 167,85 168,13 167,88 168,35 168,10 167,79 167,86 168,09 167,93 168,00 0,17 1stp 153,92 153,88 153,88 155,02 153,90 153,86 153,96 155,03 153,77 153,92 154,11 0,48 1tdb 183,40 185,66 183,00 183,44 184,47 184,90 183,94 183,59 183,79 183,09 183,93 0,85 1tng 37,67 37,27 37,29 37,68 37,28 37,26 37,47 37,33 37,32 37,33 37,39 0,16 1tnl 45,88 45,44 51,17 43,40 45,42 45,39 45,43 45,42 45,37 45,41 45,83 1,99 1tph 70,16 69,52 70,14 69,46 69,47 70,27 69,52 70,14 69,47 69,49 69,76 0,36 1tpp 173,55 173,30 174,85 173,67 174,86 174,82 173,69 173,41 173,66 173,49 173,93 0,64 1ukz 538,17 538,22 535,79 535,79 535,67 540,50 538,52 535,67 537,67 535,70 537,17 1,69 1ulb 32,23 32,58 32,23 32,24 32,50 32,80 32,59 32,22 32,49 32,52 32,44 0,20 2ack 56,07 56,68 56,09 56,81 56,71 56,07 56,69 56,08 56,08 56,10 56,34 0,33 2ak3 324,89 320,78 322,89 321,42 320,61 321,48 324,85 320,82 321,15 320,75 321,96 1,66 2cgr 397,27 400,07 400,13 399,92 399,62 435,83 399,85 399,91 403,90 402,03 403,85 11,36 2cht 79,68 80,39 79,68 79,68 80,42 79,68 79,67 80,41 79,67 79,70 79,90 0,35 2cmd 122,68 122,50 122,63 122,51 122,71 123,35 122,57 122,53 122,59 122,65 122,67 0,25 2dbl 321,30 320,77 323,54 320,96 321,50 321,42 321,37 321,33 321,37 323,46 321,70 0,97 2gbp 167,40 167,37 168,58 167,55 167,50 167,40 166,94 166,96 167,38 168,56 167,56 0,57 2r07 401,26 401,58 399,49 399,31 399,90 399,88 399,57 400,08 399,71 399,69 400,05 0,76 2sim 277,30 275,15 275,95 274,87 275,83 276,17 275,24 275,06 275,26 277,47 275,83 0,92 3hvt 73,37 73,38 73,39 74,28 74,38 74,28 74,16 73,38 73,41 73,43 73,75 0,46 3ptb 40,23 40,43 40,65 40,24 40,83 40,65 40,30 40,26 40,64 40,85 40,51 0,24 3tpi 55,33 55,67 55,64 55,34 55,35 55,67 55,38 55,42 55,34 55,36 55,45 0,15 4cts 44,51 44,83 44,51 44,86 44,83 44,51 44,88 44,52 44,51 44,83 44,68 0,18 4fab 192,22 191,34 192,76 191,08 192,89 192,73 191,31 192,73 191,35 192,78 192,12 0,75 6abp 20,62 20,27 20,59 20,30 20,30 20,58 20,29 20,26 20,30 20,28 20,38 0,15 7tim 74,54 74,57 74,58 74,54 74,55 74,55 74,39 74,39 74,52 75,37 74,60 0,28 8atc 144,39 143,58 144,41 143,49 144,53 144,21 143,48 143,60 144,28 143,63 143,96 0,44 8gch 14,44 14,31 14,30 14,32 14,30 14,41 14,31 14,41 14,30 14,39 14,35 0,06
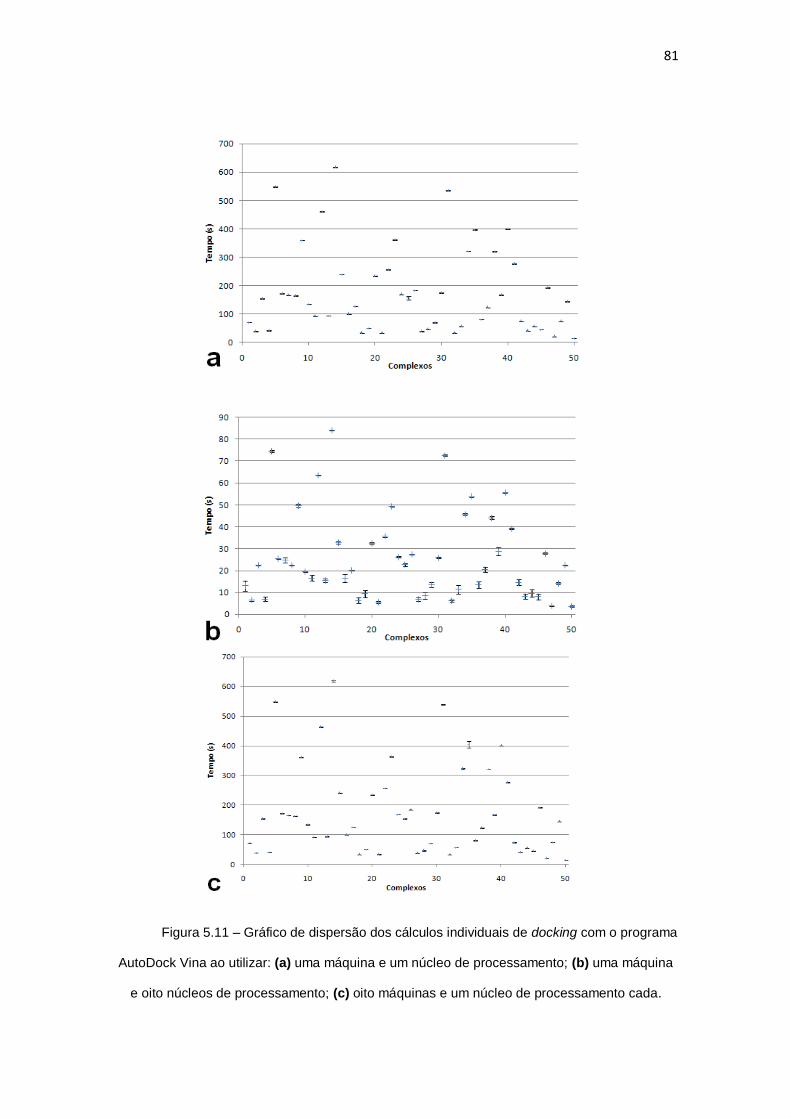
81
Figura 5.11 – Gráfico de dispersão dos cálculos individuais de docking com o programa
AutoDock Vina ao utilizar: (a) uma máquina e um núcleo de processamento; (b) uma máquina
e oito núcleos de processamento; (c) oito máquinas e um núcleo de processamento cada.

82
As pequenas barras nos gráficos da Figura 5.11 representam o desvio
padrão ao redor da média de tempo das 10 execução de cada complexo dentro
de um mesmo job, uma vez que cada job contém 10 repetições de re-docking
com cada complexo. Conforme se pode observar nestes gráficos o desvio
padrão no tempo de execução das cópias foi baixo em relação à média do
conjunto de cópias de execução de cada cálculo de docking realizado nos
complexos ligante-receptor. Isto indica que estes cálculos de docking
realizados nestas configurações (“setup”) do programa AutoDock Vina parecem
ser significativamente reprodutíveis, particularmente no que diz respeito à sua
demanda computacional. Isto também pode ser observado nos cálculos com
oito máquinas (Tabela 14) (Figura 5.11c) onde também ocorreu um desvio
padrão baixo em relação à média do conjunto de cópias de execução de cada
cálculo de docking realizado nos complexos ligante-receptor. Neste cálculo de
docking com oito máquinas há uma grande chance de que as cópias de
execução de cada cálculo tenham sido realizadas por máquinas diferentes,
reforçando ainda mais esta questão da reprodutibilidade da demanda
computacional dos cálculos ou tarefas individuais, sendo realizadas no
ambiente de grid computacional.
Acredita-se que a configuração dos parâmetros seed e exhaustiveness
tornaram o cálculo reprodutível em demanda computacional. O parâmetro seed
(semente inicial para o gerador de números aleatórios) é responsável por
determinar a maneira com que o procedimento de docking inicia sua busca. Ao
se configurar um valor fixo para este parâmetro, o algoritmo genético sempre
começa sua busca da mesma maneira e, desta forma, tende a um mesmo
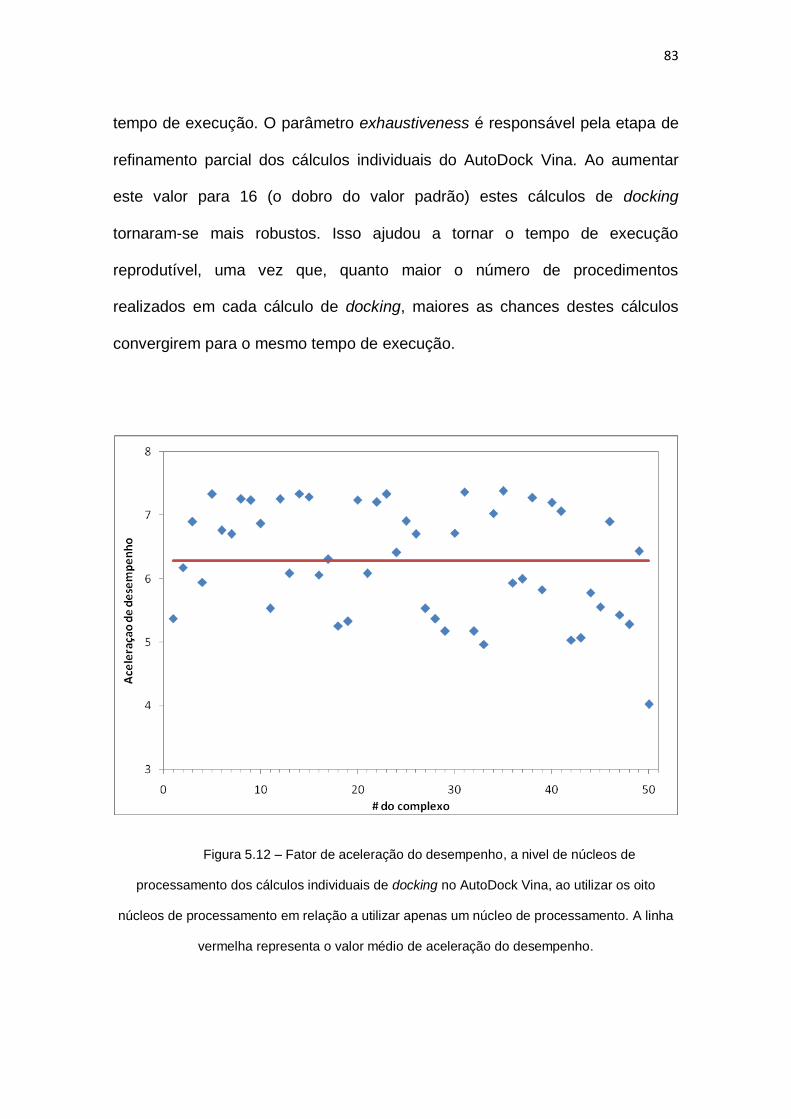
83
tempo de execução. O parâmetro exhaustiveness é responsável pela etapa de
refinamento parcial dos cálculos individuais do AutoDock Vina. Ao aumentar
este valor para 16 (o dobro do valor padrão) estes cálculos de docking
tornaram-se mais robustos. Isso ajudou a tornar o tempo de execução
reprodutível, uma vez que, quanto maior o número de procedimentos
realizados em cada cálculo de docking, maiores as chances destes cálculos
convergirem para o mesmo tempo de execução.
Figura 5.12 – Fator de aceleração do desempenho, a nivel de núcleos de
processamento dos cálculos individuais de docking no AutoDock Vina, ao utilizar os oito
núcleos de processamento em relação a utilizar apenas um núcleo de processamento. A linha
vermelha representa o valor médio de aceleração do desempenho.

84
Tomando por base o que foi observado no estudo original do AutoDock
Vina (TROTT; OLSON, 2010), onde também não foi obtida a aceleração de
desempenho ideal de oito vezes (com oito núcleos de processamento),
comparado com o cálculo utilizando apenas um núcleo de processamento. O
valor de aceleração médio com oito núcleos de processamento, relatado nesta
referência bibliográfica (TROTT; OLSON, 2010) é de 7,25 vezes (gráfico 5 da
página 459). Nos testes realizados neste presente trabalho, a aceleração do
desempenho variou entre 4,02 a 7,38 vezes mais rápido quando comparado
com o cálculo utilizando apenas um núcleo de processamento (ver Figura
5.12). Acredita-se que esta diferença em relação ao estudo do AutoDock Vina
tenha ocorrido devido a diferenças de configuração das máquinas (a nível de
software e de hardware) utilizadas neste estudo, em comparação com as
configurações utilizadas no estudo original do AutoDock Vina. Novamente,
cabe aqui ressaltar que a capacidade de paralelização interna do programa
AutoDock Vina não foi o objetivo central deste projeto de mestrado, uma vez
que este programa foi desenvolvido por terceiros, mas a principal tarefa deste
presente trabalho é medir o ganho de desempenho proveniente do emprego da
computação distribuída em um ambiente de grid computacional.
5.2.2.2 Resultados da reordenamento da sequência de tarefas no job
Com o intuito de verificar se a ordem em que os cálculos individuais de
docking são realizados influencia o tempo total de execução do job (conjunto
de cálculos), testaram-se outras formas de ordenamento destes cálculos de
docking. Estes ordenamentos foram realizados levando-se em consideração a

85
complexidade ou a demanda computacional dos cálculos, tanto em ordem
crescente quanto decrescente, além de ordens aleatórias (Figura 5.13). Os
tempos totais de execução destes jobs foram comparados com o mesmo
ordenamento utilizado para os testes padrões de desempenho (ordem
alfabética dos nomes dos complexos, ligante + receptor). Cada teste de
ordenamento foi repetido cinco vezes. Na ordem aleatória, foram gerados cinco
jobs contendo ordens aleatórias diferentes entre si. Todos os cálculos da
Tabela 15 foram realizados em oito máquinas utilizando todos os oito núcleos
de processamento de cada máquina, com o programa AutoDock Vina.
Tabela 15 – Tempos de execução dos cálculos utilizando diferentes sequências de
ordenamento das tarefas individuais (docking).
# Ordem aleatória
Ordem alfabética
Ordem crescente
Ordem decrescente
1 2513 2823 3088 3093
2 2589 2847 3089 3155
3 2863 2798 3026 3103
4 2547 2813 3068 3067
5 2523 2819 3017 3085
Média 2607 2820 3058 3101
Desvio Padrão 146 18 34 33
Como os testes em ordem alfabética já tinham sido realizados por
padrão, já havia ciência sobre a demanda computacional ou sobre a
complexidade de cada um dos complexos (ligante + receptor), portanto foi
possível fazer um ordenamento neste sentido. No ordenamento crescente de
complexidade, os cálculos com maior complexidade ficaram no final. Ao utilizar

86
esta forma de ordenamento observou-se uma perda global de desempenho
durante a execução do job (conjunto de cálculos).
Figura 5.13 – Desempenho ao reordenar a sequência dos cálculos individuais de docking. O
eixo das ordenadas representa o tempo em segundos, enquanto o eixo das abscissas
representa as 5 repetições de cada teste.
Acredita-se que neste caso, durante os últimos cálculos, as máquinas
que já concluíram seus cálculos permanecem ociosas a espera que as
máquinas restantes finalizem os últimos cálculos, que são os mais demorados
(de maior complexidade ou demanda computacional), afetando assim o
desempenho do grid computacional. Curiosamente, ao contrário do que se
esperava encontrar, o ordenamento decrescente, ou seja, através da
diminuição do nível de complexidade a cada cálculo, também tornou o conjunto

87
de cálculos um pouco mais lento, como um todo. Vale ressaltar que, como não
há como saber a demanda computacional de cada cálculo de docking antes
mesmo de executá-los, estes tipos de ordenamentos por nível de complexidade
são inviáveis de serem implementados e utilizados na prática, mesmo sabendo
que há uma correlação conhecida entre o tempo de cálculo (ou a complexidade
computacional) e o tamanho das moléculas e do sítio ativo, além do número de
graus de liberdade envolvidos.
Observa-se que, com exceção do terceiro dos cinco testes, os cálculos
com o ordenamento aleatório obtiveram um tempo de execução melhor do que
o ordenamento alfabético padrão. Desta forma, para melhor analisar o efeito do
ordenamento aleatório no desempenho dos cálculos de docking no ambiente
de grid computacional, foram gerados (utilizando os mesmos 500 cálculos de
docking do job original) mais dez jobs aleatórios distintos entre si, ou seja, em
ordens diferentes, e realizados (Tabela 16) (Figura 5.14) em uma máquina
utilizando todos os oito núcleos de processamento (M1C8) e em oito máquinas
utilizando todos os oito núcleos de processamento (M8C8).
Tabela 16 – Tempos de execução dos jobs aleatórios, em segundos.
# Job Aleatório
M1C8 M8C8
1 24110 2846
2 24078 2830
3 24045 2840
4 24102 2837
5 24111 2842
6 23977 2875
7 21402 2859
8 20948 2871
9 20381 2817
10 20630 2887
Média 22778 2850
Desvio Padrão 1688 22
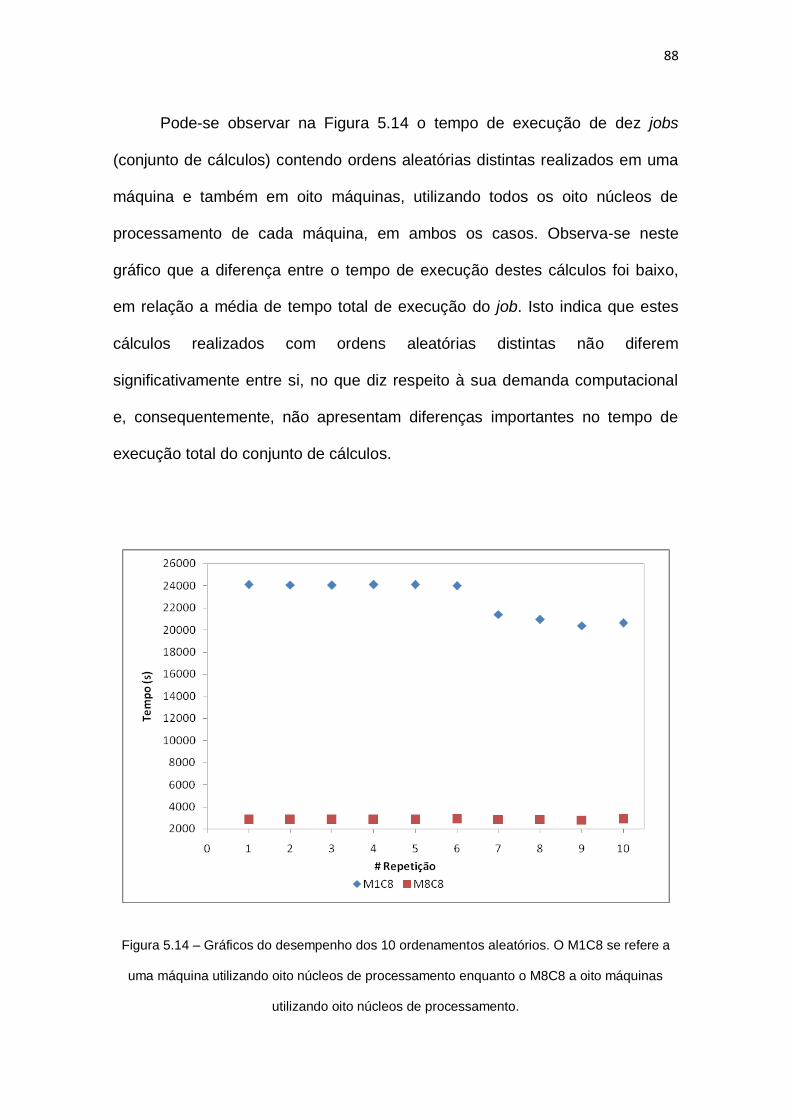
88
Pode-se observar na Figura 5.14 o tempo de execução de dez jobs
(conjunto de cálculos) contendo ordens aleatórias distintas realizados em uma
máquina e também em oito máquinas, utilizando todos os oito núcleos de
processamento de cada máquina, em ambos os casos. Observa-se neste
gráfico que a diferença entre o tempo de execução destes cálculos foi baixo,
em relação a média de tempo total de execução do job. Isto indica que estes
cálculos realizados com ordens aleatórias distintas não diferem
significativamente entre si, no que diz respeito à sua demanda computacional
e, consequentemente, não apresentam diferenças importantes no tempo de
execução total do conjunto de cálculos.
Figura 5.14 – Gráficos do desempenho dos 10 ordenamentos aleatórios. O M1C8 se refere a
uma máquina utilizando oito núcleos de processamento enquanto o M8C8 a oito máquinas
utilizando oito núcleos de processamento.

89
Com estes testes adicionais com o ordenamento aleatório, obteve-se,
em geral, um desempenho semelhante ao ordenamento alfabético. Isso se
deve ao fato que, tratando-se de complexidade, o ordenamento alfabético
também é aleatório. Estes ordenamentos não sobrecarregaram nem
subutilizaram o ambiente de grid computacional, por isso foram obtidos
melhores desempenhos globais do grid computacional usando-se estes
ordenamentos. Desta forma, pode-se observar que o balanceamento da
demanda computacional é mais benéfico para o desempenho do ambiente de
grid computacional, controlado pelo GriDoMol, do que a concentração dos
cálculos de docking baseando-se em sua complexidade.

90
6 CONCLUSÕES
Neste presente trabalho, observou-se alguns fatores que causam o
aumento de desempenho no ambiente de grid computacional. Isto se deve por
causa da ocorrência simultânea das fases de processamento de dados,
transferência de arquivos e comunicações neste sistema distribuído, mesmo
que ocorrendo em fases diferentes. Por exemplo, se uma determinada máquina
está na fase de transferência de arquivos, enquanto a outra máquina está na
fase de processamento de dados, faz com que o tempo gasto na execução em
paralelo destas fases seja contado apenas uma vez, reduzindo
consideravelmente o tempo necessário para concluir um determinado número
de tarefas em comparação com o caso em que estas fases estão sendo
realizadas sequencialmente em uma única máquina, onde o tempo gasto na
execução sequencial destas fases é sempre cumulativo.
Por outro lado, a ociosidade nas fases de processamento de dados,
transferência de arquivos, e comunicação entre os componentes representa
algumas das limitações encontradas neste tipo de sistema distribuido. A
ociosidade na fase de processamento de dados acontece quando há mais
máquinas disponíveis do que as tarefas a serem executadas. Por exemplo, ao
realizar as últimas três tarefas em um ambiente de grid computacional
contendo 10 máquinas, sete destas máquinas estarão ociososas aguardando
as máquinas restantes terminarem estas últimas tarefas. Também observa-se
ociosidade durante as fases de transferência de arquivos e comunicação no
ambiente de grid computacional, quando comparado com uma execução em

91
apenas uma máquina, umas vez que neste último caso, os arquivos já estão
disponibilizados na máquina, eliminando assim o tempo gasto com estas fases.
Mesmo levando em conta estas limitações mencionadas acima, o
aumento de desempenho obtido utilizando as estratégias de paralelismo, no
nível de máquinas do grid computacional, no nível de núcleos de
processamento (AutoDock Vina) e no nível de instâncias de execução
(AutoDock), foi de 33 e 34 vezes mais rápido do que utilizar apenas uma
máquina e um núcleo de processamento para o AutoDock e AutoDock Vina,
respectivamente. Desta forma, o tempo necessário para a realização de 500
tarefas de docking molecular foi reduzido em cerca de 97%, em comparação
com o tempo necessário quando não é utilizada qualquer estratégia de
paralelismo, ou de computação distribuída. Vale ressaltar que este ganho de
desempenho é praticamente linear e, ao adicionar mais máquinas ao ambiente
de grid computacional, é possível obter desempenhos ainda melhores de
redução de tempo total para a realização do job completo (múltiplas tarefas).
Assim, pode-se concluir que o esforço de implementação do programa
GriDoMol, permitindo a utilização dos recursos de computação distribuída
ofertados pelo ambiente de grid computacional OurGrid, em conjunto com os
programas AutoDock e AutoDock Vina, alcançou seu objetivo principal de
aumentar o desempenho computacional, permitindo a realização de um
conjunto grande de tarefas individuais (“BoT”) em uma pequena fração do
tempo (cerca de 3%) que seria necessário para realizar o mesmo conjunto de
tarefas em apenas um computador, de maneira sequencial.

92
7 PERSPECTIVAS
Embora tenha sido desenvolvida neste trabalho uma plataforma para
execução de cálculos de docking molecular de forma distribuída, há espaço
para inclusão de ferramentas de preparação de moléculas e de pós analise.
Algumas destas possibilidades são:
Contemplar mais programas para docking molecular na interface do
GriDoMol.
Utilizar mais rotinas computacionais (scripts) presentes no AutoDock Tools
para possibilitar a opção de preparação automatizada das moléculas que
serão utilizadas nos cálculos de docking molecular.
Oferecer mais opções de interação com GriDoMol através de interfaces,
como, por exemplo, aplicação web e aplicativo para plataforma Android.
Permitir a realização de analise pós-docking, utilizando ferramentas
disponíveis na maquina do usuário.
Possibilitar a realização de refinamento dos resultados, como, por exemplo,
utilizar funções de pontuação (score) alternativas para as soluções de
docking molecular, dinâmica molecular e otimização de geometria.
Possibilitar que o usuário escolha, diretamente da internet, as moléculas
(ligantes e receptores) que serão utilizadas na realização de docking
molecular, como, por exemplo, através de códigos PDB.

93
8 REFERÊNCIAS
ABREU, R. M. V.; FROUFE, H. J. C.; QUEIROZ, M.; FERREIRA, I. MOLA: a bootable, self-
configuring system for virtual screening using AutoDock4/Vina on computer clusters. Journal of
Cheminformatics, v. 2, 2010. ISSN 1758-2946.
ADT (2012). Disponível em: <http://autodock.scripps.edu/resources/adt>. Acessado em
Setembro de 2012.
ANDRADE, N.; CIRNE, W.; BRASILEIRO, F.; ROISENBERG, P. OurGrid: An approach to easily assemble grids with equitable resource sharing. In: FEITELSON, D.;RUDOLPH, L., et al (Ed.). Job Scheduling Strategies for Parallel Processing. Berlin: Springer-Verlag Berlin, v.2862, 2003. p.61-86. (Lecture Notes in Computer Science). ISBN 0302-9743
ARAUJO, E.; CIRNE, W.; WAGNER, G.; OLIVEIRA, N.; SOUZA, E. P.; GALVAO, C. O.; MARTINS, E. S. The SegHidro experience: Using the grid to empower a hydro-meteorological scientific network. Los Alamitos: Ieee Computer Soc, 2005. 64-71 ISBN 0-7695-2448-6.
AutoDock (2012). Disponível em <http://autodock.scripps.edu/>. Acessado em Novembro de 2012.
BUYYA, R.; BRANSON, K.; GIDDY, J.; ABRAMSON, D. The virtual laboratory: A toolset for utilising the world-wide grid to design drugs. Los Alamitos: Ieee Computer Soc, 2002. 278-
279 ISBN 0-7695-1582-7.
CASE, D. A.; CHEATHAM, T. E.; DARDEN, T.; GOHLKE, H.; LUO, R.; MERZ, K. M.;
ONUFRIEV, A.; SIMMERLING, C.; WANG, B.; WOODS, R. J. The Amber biomolecular
simulation programs. Journal of Computational Chemistry, v. 26, n. 16, p. 1668-1688, Dec
2005. ISSN 0192-8651.
Choosing BOINC projects (2012). Disponível em <http://boinc.berkeley.edu/projects.php>.
Acessado em Novembro de 2012.
CIRNE, W. (2002). Grids Computacionais Arquiteturas, Tecnologias e Aplicações. In Proceedings of the 3rd Workshop on High Performance Computing Systems (WSCAD’2002), Vitória, Brazil.
CUDA (2012). Disponível em <http://www.nvidia.com/object/cuda_home_new.html>. Acessado em Novembro de 2012.
HTCondor (2013). Disponível em <http://research.cs.wisc.edu/htcondor/>. Acessado em
Fevereiro de 2013.
DE VRIES, S. J.; VAN DIJK, M.; BONVIN, A. The HADDOCK web server for data-driven biomolecular docking. Nature Protocols, v. 5, n. 5, p. 883-897, 2010. ISSN 1754-2189.
DELANO, W. L. Use of PYMOL as a communications tool for molecular science. Abstracts of Papers of the American Chemical Society, v. 228, p. U313-U314, Aug 2004. ISSN 0065-7727.
DELANO, W. L. PyMOL molecular viewer: Updates and refinements. Abstracts of Papers of the American Chemical Society, v. 238, Aug 2009. ISSN 0065-7727.

94
DESJARLAIS, R. L.; SEIBEL, G. L.; KUNTZ, I. D.; FURTH, P. S.; ALVAREZ, J. C.; DEMONTELLANO, P. R. O.; DECAMP, D. L.; BABE, L. M.; CRAIK, C. S. STRUCTURE-BASED DESIGN OF NONPEPTIDE INHIBITORS SPECIFIC FOR THE HUMAN IMMUNODEFICIENCY VIRUS-1 PROTEASE. Proceedings of the National Academy of Sciences of the United States of America, v. 87, n. 17, p. 6644-6648, Sep 1990. ISSN 0027-8424.
DOMINGUEZ, C.; BOELENS, R.; BONVIN, A. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society,
v. 125, n. 7, p. 1731-1737, Feb 2003. ISSN 0002-7863.
DURRANT, J. D.; MCCAMMON, J. A. BINANA: A novel algorithm for ligand-binding characterization. Journal of Molecular Graphics & Modelling, v. 29, n. 6, p. 888-893, Apr 2011. ISSN 1093-3263.
e-NMR (2012). Disponível em: <http://www.enmr.eu/>. Acessado em Outubro de 2012.
Ejabberd (2012). Disponível em: <http://www.ejabberd.im/>. Acessado em Outubro de 2012.
FISCHER, E. (1894). "Einfluss der Configuration auf die Wirkung der Enzyme". Ber. Dt.
Chem. Ges. 27: 2985–2993.
FOSTER, I. (2002). What is the grid? A three point checklist.
GELDENHUYS, W. J.; GAASCH, K. E.; WATSON, M.; ALLEN, D. D.; VAN DER SCHYF, C. J. Optimizing the use of open-source software applications in drug discovery. Drug Discov Today, v. 11, n. 3-4, p. 127-32, Feb 2006. ISSN 1359-6446
GIUPPONI, G.; HARVEY, M. J.; DE FABRITIIS, G. The impact of accelerator processors for high-throughput molecular modeling and simulation. Drug Discovery Today, v. 13, n. 23-24, p.
1052-1058, Dec 2008. ISSN 1359-6446.
GOODSELL, D. S.; MORRIS, G. M.; OLSON, A. J. Automated docking of flexible ligands: Applications of AutoDock. Journal of Molecular Recognition, v. 9, n. 1, p. 1-5, Jan-Feb 1996. ISSN 0952-3499.
HESS, B.; KUTZNER, C.; VAN DER SPOEL, D.; LINDAHL, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, v. 4, n. 3, p. 435-447, Mar 2008. ISSN 1549-9618.
JIANG, X. H.; KUMAR, K.; HU, X.; WALLQVIST, A.; REIFMAN, J. DOVIS 2.0: an efficient and easy to use parallel virtual screening tool based on AutoDock 4.0. Chemistry Central Journal, v. 2, Sep 2008. ISSN 1752-153X.
KELLENBERGER, E.; RODRIGO, J.; MULLER, P.; ROGNAN, D. Comparative evaluation of
eight docking tools for docking and virtual screening accuracy. Proteins-Structure Function
and Bioinformatics, v. 57, n. 2, p. 225-242, Nov 2004. ISSN 0887-3585.
KOSHLAND, D. E. APPLICATION OF A THEORY OF ENZYME SPECIFICITY TO PROTEIN
SYNTHESIS. Proceedings of the National Academy of Sciences of the United States of
America, v. 44, n. 2, p. 98-104, 1958. ISSN 0027-8424.
LAM/MPI Parallel Computing (2012). Disponível em <http://www.lam-mpi.org/>. Acessado em
Novembro de 2012.

95
LENGAUER, T.; RAREY, M. Computational methods for biomolecular docking. Current Opinion in Structural Biology, v. 6, n. 3, p. 402-406, Jun 1996. ISSN 0959-440X.
Linpack (2013). Disponível em <http://www.netlib.org/linpack/>. Acessado em Fevereiro de
2013.
MATTOS, G.; FORMIGA, A. D.; LINS, R. D.; MARTINS, F. M. J.; ACM. BigBatch: A
Document Processing Platform for Clusters and Grids. New York: Assoc Computing
Machinery, 2008. 434-441
MCGEE, P. (2005). Modeling success with in silico tools. Drug Discovery & Development. 8,
24.
MDL Isis Draw (2012). Disponível em: <http://mdl-isis-draw.software.informer .com/>. Acessado
em Outubro de 2012.
Mingw (2013). Disponível em: <www.mingw.org/>. Acessado em Fevereiro de 2013.
MORRIS, G. M.; HUEY, R.; LINDSTROM, W.; SANNER, M. F.; BELEW, R. K.; GOODSELL, D.
S.; OLSON, A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor
Flexibility. Journal of Computational Chemistry, v. 30, n. 16, p. 2785-2791, Dec 2009. ISSN
0192-8651.
MPICH2 : High-performance and widely portable MPI (2012). Disponível em
<http://www.mcs.anl.gov/research/projects/mpich2/>. Acessado em Novembro de 2012.
NSF - National Science Foundation (2012). Disponível em: <http://www.nsf.gov/>. Acessado
em Outubro de 2012.
OpenFire (2012). Disponível em: <http://www.igniterealtime.org/projects/ openfire/>. Acessado
em Outubro de 2012.
OurGrid (2012). Disponível em: <http://www.ourgrid.org/>. Acessado em Outubro de 2012.
OurGrid Portal (2012). Disponível em: <http://portal.ourgrid.org/>. Acessado em Outubro de
2012.
PAPPALARDO, F.; HALLING-BROWN, M. D.; RAPIN, N.; ZHANG, P.; ALEMANI, D.;
EMERSON, A.; PACI, P.; DUROUX, P.; PENNISI, M.; PALLADINI, A.; MIOTTO, O.;
CHURCHILL, D.; ROSSI, E.; SHEPHERD, A. J.; MOSS, D. S.; CASTIGLIONE, F.;
BERNASCHI, M.; LEFRANC, M. P.; BRUNAK, S.; MOTTA, S.; LOLLINI, P. L.; BASFORD, K.
E.; BRUSIC, V. ImmunoGrid, an integrative environment for large-scale simulation of the
immune system for vaccine discovery, design and optimization. Briefings in Bioinformatics, v.
10, n. 3, p. 330-340, May 2009. ISSN 1467-5463.
PDB Database (2012). Disponível em: <http://www.pdb.org>. Acessado em Outubro de 2012.

96
PEARLMAN, D. A.; CASE, D. A.; CALDWELL, J. W.; ROSS, W. S.; CHEATHAM, T. E.; DEBOLT, S.; FERGUSON, D.; SEIBEL, G.; KOLLMAN, P. AMBER, A PACKAGE OF COMPUTER-PROGRAMS FOR APPLYING MOLECULAR MECHANICS, NORMAL-MODE ANALYSIS, MOLECULAR-DYNAMICS AND FREE-ENERGY CALCULATIONS TO SIMULATE THE STRUCTURAL AND ENERGETIC PROPERTIES OF MOLECULES. Computer Physics Communications, v. 91, n. 1-3, p. 1-41, Sep 1995. ISSN 0010-4655.
PURANEN, J. S.; VAINIO, M. J.; JOHNSON, M. S. Accurate Conformation-Dependent Molecular Electrostatic Potentials for High-Throughput In Silico Drug Discovery. Journal of Computational Chemistry, v. 31, n. 8, p. 1722-1732, Jun 2010. ISSN 0192-8651.
QT – A cross-platform aplication and UI framework (2012). Disponível em:
<http://qt.digia.com/>. Acessado em Julho de 2012.
ROH, Y.; LEE, J.; PARK, S.; KIM, J.-I. A molecular docking system using CUDA.
Proceedings of the 2009 International Conference on Hybrid Information Technology. Daejeon, Korea: ACM: 28-33 p. 2009.
SADASHIV, N.; KUMAR, S. M. D. Cluster, grid and cloud computing: A detailed comparison. Computer Science & Education (ICCSE), 2011 6th International Conference on, 2011. 3-5 Aug. 2011. p.477-482.
SAYLE, R. A.; MILNERWHITE, E. J. RASMOL - BIOMOLECULAR GRAPHICS FOR ALL. Trends in Biochemical Sciences, v. 20, n. 9, p. 374-376, Sep 1995. ISSN 0968-0004.
SCHOLLMEIER, R. A definition of Peer-to-Peer networking for the classification of Peer-to-Peer architectures and applications. Los Alamitos: Ieee Computer Soc, 2002. 101-102 ISBN 0-7695-1503-7.
Scripps Research Institute (2012). Disponível em: <http://www.scripps.edu/>. Acessado em
Outubro de 2012.
SLOCCount (2012). Disponível em <http://www.dwheeler.com/sloccount/>. Acessado em
Novembro de 2012.
SLOCCount User’s Guide (2012). Disponível em <http://www.dwheeler.com/sloccount/
sloccount.html>. Acessado em Novembro de 2012.
SMITH, R. Grid Computing: A Brief Technology Analysis.
SQUYRES, J. M.; LUMSDAINE, A. A component architecture for LAM/MPI. In: DONGARRA, J.;LAFORENZA, D., et al (Ed.). Recent Advances in Parallel Virtual Machine and Message Passing Interface. Berlin: Springer-Verlag Berlin, v.2840, 2003. p.379-387. (Lecture Notes in
Computer Science). ISBN 0302-9743.
SUKHWANI, B.; HERBORDT, M. C. Fast binding site mapping using GPUs and CUDA. Parallel & Distributed Processing, Workshops and Phd Forum (IPDPSW), 2010 IEEE International Symposium on, 2010. 19-23 April 2010. p.1-8.
The Linux Kernel: It’s Worth More (2012). Disponível em: <http://www.
dwheeler.com/essays/linux-kernel-cost.html>. Acessado em Novembro de 2012.
TOP500 Supercomputers (2012). Disponível em <www.top500.org/>. Acessado em Novembro
de 2012.

97
TROTT, O.; OLSON, A. J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry, v. 31, n. 2, p. 455-461, Jan 2010. ISSN
0192-8651.
VAINIO, M. J.; JOHNSON, M. S. Generating conformer ensembles using a multiobjective genetic algorithm. Journal of Chemical Information and Modeling, v. 47, n. 6, p. 2462-2474, Nov-Dec 2007. ISSN 1549-9596.
VAQUE, M.; ARDREVOL, A.; BLADE, C.; SALVADO, M. J.; BLAY, M.; FERNANDEZ-LARREA, J.; AROLA, L.; PUJADAS, G. Protein-ligand docking: A review of recent advances and future perspectives. Current Pharmaceutical Analysis, v. 4, n. 1, p. 1-19, Feb 2008. ISSN 1573-4129.
WLODAWER, A. Rational approach to AIDS drug design through structural biology. Annual Review of Medicine, v. 53, p. 595-614, 2002. ISSN 0066-4219.
WOODS, C. J.; NG, M. H.; JOHNSTON, S.; MURDOCK, S. E.; WU, B.; TAI, K.; FANGOHR, H.;
JEFFREYS, P.; COX, S.; FREY, J. G.; SANSOM, M. S. P.; ESSEX, J. W. Grid computing and
biomolecular simulation. Philosophical Transactions of the Royal Society a-Mathematical
Physical and Engineering Sciences, v. 363, n. 1833, p. 2017-2035, Aug 2005. ISSN 1364-
503X.
World Community Grid (2012). Disponível em <http://www.worldcommunitygrid. org>. Acessado
em Outubro de 2012.
World Community Grid – Say No to Schistosoma (2012). Disponível em <http://
www.worldcommunitygrid.org/research/sn2s/overview.do>. Acessado em Outubro de 2012.
World Community Grid - GO Fight Against Malaria (2012). Disponível em <http:
//www.worldcommunitygrid.org/research/gfam/overview.do>. Acessado em Outubro de 2012.
World Community Grid - Drug Search for Leishmaniasis (2012). Disponível em
<http://www.worldcommunitygrid.org/research/dsfl/overview.do>. Acessado em Outubro de
2012.
XDrawChem (2012). Disponível em: <http://xdrawchem.sourceforge.net/>. Acessado em
Outubro de 2012.
XSede (2012). Disponível em: <https://www.xsede.org/>. Acessado em Outubro de 2012.
ZHANG, S. X.; KUMAR, K.; JIANG, X. H.; WALLQVIST, A.; REIFMAN, J. DOVIS: an
implementation for high-throughput virtual screening using AutoDock. Bmc Bioinformatics, v.
9, Feb 2008. ISSN 1471-2105.

98
9 APENDICE A
job : label : Autogrid
task :
init : if (os == windows) then store autogrid4.exe autogrid4.exe store autogrid.bat autogrid.bat endif if (os == linux) then store autogrid4 autogrid4 store autogrid.sh autogrid.sh endif put C:/LQTM/mol/receptors/1ABE.pdbqt 1ABE.pdbqt put C:/LQTM/mol/receptors/1ABE.gpf 1ABE.gpf
remote : autogrid.sh $STORAGE 1ABE.gpf
final : get maps.zip 1ABE_maps.zip
get log_grid.txt 1ABE_autogrid_log.txt
task :
( . . . )
Exemplo resumido de um job description file (jdf) gerado para a etapa do AutoGrid.
job : label : 1x_autodock
task : init : if (os == windows) then store autodock4.exe autodock4.exe store autodock.bat autodock.bat endif if (os == linux) then store autodock4 autodock4 store autodock.sh autodock.sh endif put 1ABE_maps.zip maps.zip put C:/LQTM/mol/receptors/1ABE.pdbqt 1ABE.pdbqt put C:/LQTM/mol/ligands/1ABE-lig.pdbqt 1ABE-lig.pdbqt put C:/LQTM/mol/ligands/1ABE-lig_1ABE.dpf 1ABE-lig_1ABE.dpf
remote : autodock.sh $STORAGE 1ABE-lig_1ABE.dpf
final : get log_dock.dlg 1ABE-lig_1ABE1.dlg
task :
( . . . )
Exemplo resumido de um job description file (jdf) gerado para a execução do programa
AutoDock no ambiente de grid computacional utilizando uma instancia de execução do
programa.

99
job : label : 8x_autodock task : init : if (os == windows) then store autodock4.exe autodock4.exe store 8autodock.bat 8autodock.bat endif if (os == linux) then store autodock4 autodock4 store 8autodock.sh 8autodock.sh endif put 1ABE_maps.zip maps1.zip put C:/LQTM/mol/receptors/1ABE.pdbqt 1ABE.pdbqt put C:/LQTM/mol/ligands/1ABE-lig.pdbqt 1ABE-lig.pdbqt put 1ABE-lig_1ABE.dpf 1ABE-lig_1ABE.dpf put 1ACJ_maps.zip maps2.zip put C:/LQTM/mol/receptors/1ACJ.pdbqt 1ACJ.pdbqt put C:/LQTM/mol/ligands/1ACJ-lig.pdbqt 1ACJ-lig.pdbqt put 1ACJ-lig_1ACJ.dpf 1ACJ-lig_1ACJ.dpf put 1ACM_maps.zip maps3.zip put C:/LQTM/mol/receptors/1ACM.pdbqt 1ACM.pdbqt put C:/LQTM/mol/ligands/1ACM-lig.pdbqt 1ACM-lig.pdbqt put 1ACM-lig_1ACM.dpf 1ACM-lig_1ACM.dpf put 1AHA_maps.zip maps4.zip put C:/LQTM/mol/receptors/1AHA.pdbqt 1AHA.pdbqt put C:/LQTM/mol/ligands/1AHA-lig.pdbqt 1AHA-lig.pdbqt put 1AHA-lig_1AHA.dpf 1AHA-lig_1AHA.dpf put 1CBS_maps.zip maps5.zip put C:/LQTM/mol/receptors/1CBS.pdbqt 1CBS.pdbqt put C:/LQTM/mol/ligands/1CBS-lig.pdbqt 1CBS-lig.pdbqt put 1CBS-lig_1CBS.dpf 1CBS-lig_1CBS.dpf put 1COM_maps.zip maps6.zip put C:/LQTM/mol/receptors/1COM.pdbqt 1COM.pdbqt put C:/LQTM/mol/ligands/1COM-lig.pdbqt 1COM-lig.pdbqt put 1COM-lig_1COM.dpf 1COM-lig_1COM.dpf put 1DBB_maps.zip maps7.zip put C:/LQTM/mol/receptors/1DBB.pdbqt 1DBB.pdbqt put C:/LQTM/mol/ligands/1DBB-lig.pdbqt 1DBB-lig.pdbqt put 1DBB-lig_1DBB.dpf 1DBB-lig_1DBB.dpf put 1EED_maps.zip maps8.zip put C:/LQTM/mol/receptors/1EED.pdbqt 1EED.pdbqt put C:/LQTM/mol/ligands/1EED-lig.pdbqt 1EED-lig.pdbqt put 1EED-lig_1EED.dpf 1EED-lig_1EED.dpf remote : 8autodock.sh $STORAGE 1ABE-lig_1ABE.dpf 1ACJ-lig_1ACJ.dpf 1ACM-lig_1ACM.dpf 1AHA-lig_1AHA.dpf 1CBS-lig_1CBS.dpf 1COM-lig_1COM.dpf 1DBB-lig_1DBB.dpf 1EED-lig_1EED.dpf
final : get log_dock1.dlg 1ABE-lig_1ABE1.dlg get log_dock2.dlg 1ACJ-lig_1ACJ1.dlg get log_dock3.dlg 1ACM-lig_1ACM1.dlg get log_dock4.dlg 1AHA-lig_1AHA1.dlg get log_dock5.dlg 1CBS-lig_1CBS1.dlg get log_dock6.dlg 1COM-lig_1COM1.dlg get log_dock7.dlg 1DBB-lig_1DBB1.dlg get log_dock8.dlg 1EED-lig_1EED1.dlg task :
( . . . )
Exemplo resumido de um job description file (jdf) gerado para a execução do programa
AutoDock no ambiente de grid computacional utilizando oito instancias de execução do
programa.

100
#!/bin/bash
chmod +x $1/autogrid4
($1/autogrid4 -l log_grid.txt -p $2) && wait
zip maps.zip *.map *.maps.*
Rotina computacional em Shell Script para a execução do AutoGrid.
#!/bin/bash
chmod +x maps.zip
unzip -o maps.zip
chmod +x $1/autodock4
$1/autodock4 -l log_dock.dlg -p $2 &
for pid in `jobs -p`
do
wait $pid
done
Rotina computacional em Shell Script para a execução de uma instancia de execução do
programa AutoDock em cada máquina.

101
#!/bin/bash
chmod +x maps1.zip
chmod +x maps2.zip
( . . . )
chmod +x maps8.zip
unzip -o maps1.zip
unzip -o maps2.zip
( . . . )
unzip -o maps8.zip
chmod +x $1/autodock4
$1/autodock4 -l log_dock1.dlg -p $2 & $1/autodock4 -l log_dock2.dlg -p $3 & $1/autodock4 -l log_dock3.dlg -p $4 & $1/autodock4 -l log_dock4.dlg -p $5 & $1/autodock4 -l log_dock5.dlg -p $6 & $1/autodock4 -l log_dock6.dlg -p $7 & $1/autodock4 -l log_dock7.dlg -p $8 & $1/autodock4 -l log_dock8.dlg -p $9 &
for pid in `jobs -p`
do
wait $pid
done
Rotina computacional em Shell Script para a execução de oito instancias de execução do
programa AutoDock simultaneamente em cada máquina.

102
job : label : vina task : init : if (os == windows) then store vina.exe vina.exe endif if (os == linux) then store vina vina endif put C:/ourgrid/out/1ABE/1ABE.pdbqt 1ABE.pdbqt
put C:/LQTM/mol1ABE/1ABE-lig.pdbqt 1ABE-lig.pdbqt
remote : time -p $STORAGE/vina --receptor 1ABE.pdbqt --ligand 1ABE-lig.pdbqt --cpu 8 --seed 495829104 --exhaustiveness 16 --center_x 13.208 --center_y 58.246 --center_z 52.173 --size_x 12 --size_y 12 --size_z 12 --out 1ABE-1ABE-lig-1.out 2> 1ABE-1.log
final : get 1ABE-1ABE-lig-1.out 1ABE-1ABE-lig-1.out
get 1ABE-1.log 1ABE-1.log
task :
( . . .)
Exemplo resumido de um job description file (jdf) gerado para a execução do programa
AutoDock Vina no ambiente de grid computacional.
# This file was generated automatically by GriDoMol.
[Receptors] #path; center_x; center_y; center_z; size_x; size_y; size_z; C:/LQTM/mol/receptors/1ABE.pdbqt;13.208;58.246;52.173;12;12;12; C:/LQTM/mol/receptors/1ACJ.pdbqt;4.226;69.901;65.807;10;8;12; ( . . .)
C:/Dropbox/LQTM/mol/receptors/8GCH.pdbqt;3.092;8.142;17.699;14;14;14;
[Ligands] C:/LQTM/mol/ligands/1ABE-lig.pdbqt C:/LQTM/mol/ligands/1ACJ-lig.pdbqt
( . . . )
C:/LQTM/mol/ligands/8GCH-lig.pdbqt
Exemplo resumido do GriDoMol Settings File (gsf).

103
10 APENDICE B
13-Feb-2013
Dear Prof. Dr. Hernandes:
Your manuscript has been successfully submitted to the
Journal of Chemical Information and Modeling.
Title: "GriDoMol: a multilevel distributed system to speed
up molecular docking calculations"
Authors: Ferreira, Luiz Felipe; Cavalcante, Klaus;
Hernandes, Marcelo
Manuscript ID: ci-2013-001037.
Please reference the above manuscript ID in all future
correspondence or when calling the office for questions. If
there are any changes in your contact information, please
log in to ACS Paragon Plus at
https://acs.manuscriptcentral.com/acs and select "Edit Your
Account" to update that information.
You can view the status of your manuscript by checking your
"Authoring Activity" tab on ACS Paragon Plus after logging
in to https://acs.manuscriptcentral.com/acs.
Thank you for submitting your manuscript to the Journal of
Chemical Information and Modeling.
Sincerely,
Journal of Chemical Information and Modeling Editorial
Office

104
GriDoMol: a multilevel distributed system to
speed up molecular docking calculations
Luiz Felipe G. R. Ferreira, Klaus R. Cavalcante, Marcelo Z. Hernandes*
Department of Pharmaceutical Sciences, Federal University of Pernambuco, Brazil
KEYWORDS:
GriDoMol, distributed computing, computational grids, molecular docking, software.
ABSTRACT:
The use of computers related to drug development in the pharmaceutical area has taken
significant proportions. It is estimated that the use of these methods can reduce the costs
and time spent in the development stage of a new drug up to 50%. This often happens
because the number of molecules that need to be experimentally synthesized and tested
becomes drastically reduced due to the high predictability and reliability of the
theoretical predictions made by computational methods (in silico). The molecular
docking procedure determines if there is favorable interaction energy between two
molecules (ligand and biological or pharmacological target) in order to elucidate the
molecular reasons responsible for the pharmacological potency of these potential drugs.
It happens that these methods may present, sometimes, a high computational demand
when the number of ligands and targets to be tested is high and when there is a need for
higher precision in the predicted numerical results. This work presents the GriDoMol

105
system that performs molecular docking calculations in a computational grid
environment. Its intuitive graphical interface allows the creation of a list of molecular
docking calculations to be performed in parallel at the grid computing environment and
track the progress of each calculation individually. In tests carried out in this analysis,
the time required to perform 500 docking calculations was reduced by up to 97% of the
time required when compared to not using any parallelism strategy or distributed
computing.
Introduction
The process of drug development has acquired a growing number of computational
tools to increase the speed and decrease the costs involved in developing products
related to therapeutic innovation. The use of computers in this area has taken significant
contribution, particularly when applied on Computer-Aided Drug Design (CADD),
where structure and molecular properties are virtually studied. It is estimated that these
methods can reduce the costs and time for the development of a new drug by up to 50%
[1,2]. This happens because, usually, the number of molecules that need to be
synthesized and tested experimentally becomes drastically reduced due to the high
predictability and reliability of computational (in silico) methods, thus shortening the
development time of a new drug.
One of these methods is the molecular docking, that determinates if there is a favorable
interaction between two molecules in order to elucidate the molecular factors that are
responsible for the pharmacological activity of a new potential drug. The docking
procedure searches for a position and orientation that maximizes these intermolecular
interactions between ligands (potential drug molecules) and biological targets
(biomacromolecules, typically). Thus, both molecules (ligand and biological target, also

106
known as receptor) form a complex by structural complementarity and energetic
stabilization, as seen in Figure 1. Nevertheless, these methods present a high
computational demand when used in a huge number of molecules (ligands and
biological targets) to be tested, and also when numerical results with better accuracy are
desired.
Figure 1. Example of a structural complex (PDB code: 1ULB) formed by a receptor
(orange molecule) and its co-crystallized ligand (white molecule). The blue molecule
represents the resulting pose obtained from the molecular docking calculation. (a) A

107
panoramic view with the receptor’s surface, including the co-crystallized ligand and the
docking solution. (b) A detailed view of the receptor’s binding site.
The idea of applying distributed computing technologies, such as computational grids to
assemble virtual laboratories is currently used worldwide in projects of several research
fields [3,4,5], including molecular modeling [6,7]. In this context, it is possible to find
initiatives of studies involving molecular docking being accelerated by powerful
computing infrastructures [8,9,10,11].
There are some examples of projects that use distributed computing to perform
molecular docking in large scale. The Dovis [9] is a system that uses a computer cluster
to perform molecular docking calculations using the AutoDock [12] software. Also,
there is the Haddock [10], which is a computer cluster system that accesses the e-NMR
[13] European grid if more computational cycles are required beyond its computational
processing capacity.
Another example of distributed molecular docking is the MOLA [11] system. It also
uses a computer cluster so as to run molecular docking to large datasets. This system is
assembled through a customized LIVE-CD. It also requires that the other machines
(slave nodes) have the Preboot eXecution Environment (PXE) in order to be booted
from the front end machine (master node). This system uses the AutoDock [12] and
AutoDock Vina [14] molecular docking softwares.
In this way, the total time required for obtaining the final results of this kind of
applications can be significantly reduced, since each molecular docking calculation is
distributed within the computational grid, in a transparent way to the end user, who will

108
have the feeling that the whole process is running on a single machine, while the
computational grid is in charge of distributing and running them optimally through the
integrated system. In order to obtain a unified and intuitive platform able to perform
molecular docking in a distributed environment, this work presents GriDoMol, a
solution that integrates the processing power of a computational grid environment with
an open source and multi-core software for molecular docking. In this paper, it is
presented how much the performance on molecular docking is increased when a system
as GriDoMol, with two levels of distribution (first level at the grid machines schedule
and the second level at the processing cores) is used to perform molecular docking of a
large set of molecules.
Methods
GriDoMol is an integrated system formed by (i) a management module that has the
same name of the solution, (ii) the components (Peer, Broker and Worker) of the
OurGrid platform (v4.2.3) [15], (iii) Openfire server (v3.7.0) [16], a XMPP server used
by OurGrid to provide communication among its components, and (iv) AutoDock Vina
(v1.1.2) [14], a well-known open-source software for molecular docking.
The GriDoMol module was developed using C++ programming language and Qt toolkit
(v4.7.0) [17] to provide a friendly user interface (see Figure 2). This module is
responsible for creating the jobs, submitting them to the grid environment and
monitoring their execution. These jobs are created and exported by using the Job
Definition File (JDF) syntax, which is the file format used to submit jobs in the OurGrid
platform. Each task of the job matches a molecular docking calculation of a pair of
molecules (ligand and receptor), which also includes required input parameters and
information related to the receptor’s binding site.

109
The jobs exported by GriDoMol are scheduled among the grid machines (computers
running the component Worker of the OurGrid platform) that execute the individual
tasks (molecular docking calculations), using the AutoDock Vina software. Since Qt
framework supports the compilation on multiple operational systems, the GriDoMol
software has versions for both Windows and Linux systems.
Figure 2. Main window of the GriDoMol program.
The communication scheme among the components of the GriDoMol system can be
seen in Figure 3. The execution flow starts with an “initial setup” that includes the
preparation of the chosen molecules (ligands and targets) and the conversion of the
input data to the PDBQT file format, adopted by the AutoDock Vina program. All these
initial procedures over the molecules are made by the user with the aid of the AutoDock
Tools [12] software.

110
Figure 3. Communication scheme among the components of the GriDoMol system.
Through the graphical interface of GriDoMol, it’s possible to do the following actions:
i) Set the group of machines that integrates the computational grid by sending the Site
Description File (.SDF) to the Peer component; ii) Create and load a previously created
GriDoMol’s Setup File (.GSF), containing the setup for both receptors and ligands
molecules; iii) Create and submit a Job Description File (.JDF) to the Broker
component. This file contains the tasks of molecular docking calculations to be
performed among the computational grid machines.
After the job submission to the Broker component, it distributes the tasks optimally
through the Worker components available in the computational grid. This availability
checking is performed by the Peer component, which verifies and then selects the idle
resources to the Broker component. Each of these Workers performs the molecular
docking calculation using the AutoDock Vina program. When the task is over, each
Worker sends the results back to the user’s machine. GriDoMol also monitors, in real

111
time, which tasks have been finished and which are still running, giving an overall
progression of the job.
The OurGrid platform [15] was chosen for this work because it is suitable for the
execution of Bag-of-Tasks (BoT) applications, where the tasks have no logical or
temporal dependence among themselves, which is the case of molecular docking
calculations. Also, OurGrid is in continuous development and is being used on others
projects of high performance distributed computing and has a wide range of
applicability [3,4]. All communications among the OurGrid components are made by
the XMPP server Openfire [16]. This server detects and manages the “presence” of the
OurGrid components (if a given component is online and available) allowing the
message exchange among them.
The software used to perform the molecular docking calculations was the AutoDock
Vina [14], due to its support for multiprocessing execution in multiple CPU cores
(which is available on multi-core processors) and higher ease of use if compared to
other available docking solutions. The AutoDock Vina only requires the molecular
structures on the PDBQT file format (for both receptor and ligand) and the binding site
coordinates, both in the three-dimensional space.
The quality of performance results depends directly on the reproducibility of individual
docking calculations. So, this study attempted to map the parameters used by AutoDock
Vina that would influence the reproduction of docking calculations. We concluded that
the AutoDock Vina parameters ‘seed’ and ‘exhaustiveness’ made the calculation
reproducible in terms of computational demand. The seed parameter (initial seed for the
random number generator) is responsible for determining the way in which the docking
procedure begins its search. A fixed value for this parameter was used in all the

112
calculations. The exhaustiveness parameter is responsible for the partial refinement step
of individual calculations within AutoDock Vina. When this value is increased to 16
(twice as much the standard value) these docking calculations become more robust than
usual. These setup adjustments helped to make the execution time reproducible over all
the computations of the docking results.
Results
Comparative tests have been conducted to the proposed grid system in order to measure
the computational performance of running a large set of molecular docking calculations
primarily over the computational grid environment and also taking into account the
CPU multi-core processing, available through the use of AutoDock Vina software.
A grid environment was assembled in our lab using the OurGrid toolkit. The front-end
machine, running GriDoMol to submit and manage the jobs, was an Intel Xeon 2.80
GHz Dual-Core containing 2GB of RAM and running Windows XP. The tasks were
performed in parallel at the homogeneous grid environment, where each grid machine
had two Intel Xeon E5410 Quad-Core 2.33 GHz processors, with 16GB of RAM and
running Ubuntu Linux 10.10.
It was chosen, for performance tests, a set of molecules used by the structural
chemogenomics research group of the Strasbourg University [18]. From the original set,
a subgroup of 50 complexes (receptors and their respective ligands) was selected to
perform the docking calculation tests. The selection of these complexes was done by
removing the complexes that had metal ion or a high number of torsions on the ligand.
The complete list of ligand-target complexes and their respective PDB codes can be
seen in the Table S1 (supplementary material).

113
The impact evaluation over the performance caused by the variation in the number of
machines on the computational grid was carried out through changing the amount of
computers (Workers) involved in the dimensioning of the grid. The number of machines
used for the tests was alternated from 1 to 8 machines so as to try to determine the
performance gain on the computational grid system when more machines are added.
There was also a variation in the level of processing cores used to determine the
multicore impact. Since each grid machine has a total of 8 cores, we used a geometrical
progression (1, 2, 4 and 8 cores) as evaluation parameter, as it’s expected the double of
performance compared to the previous one.
Taking into account the number of machines of the used computational grid and the
need for higher accuracy on the tests, it was included, on each job, ten copies of the 50
complexes generating a total of 500 re-docking tasks per job. Through the possible
combination of configurations among machines (1, 2, 3, 4, 5, 6, 7 and 8) and processor
cores (1, 2, 4 and 8), 32 jobs were executed, totaling 16,000 individual re-docking
calculations tasks carried out within the computational grid. Before each docking
calculation, it was observed a systematic delay of about 10 seconds between the
command used to send the calculation (task) by the GriDoMol program interface, and
the beginning of this calculation at the work machine (Worker). This delay is typically
viewed in distributed systems that use grid architecture and is usually explained by the
initial communications between the components of the grid. The scheduling and
execution of each task within the job is preceded by the discovery and evaluation of
available resources (machines), that is, whether these machines are turned on and
connected to the grid environment, and checking the presence of the minimum
requirements needed to perform the scheduled tasks.
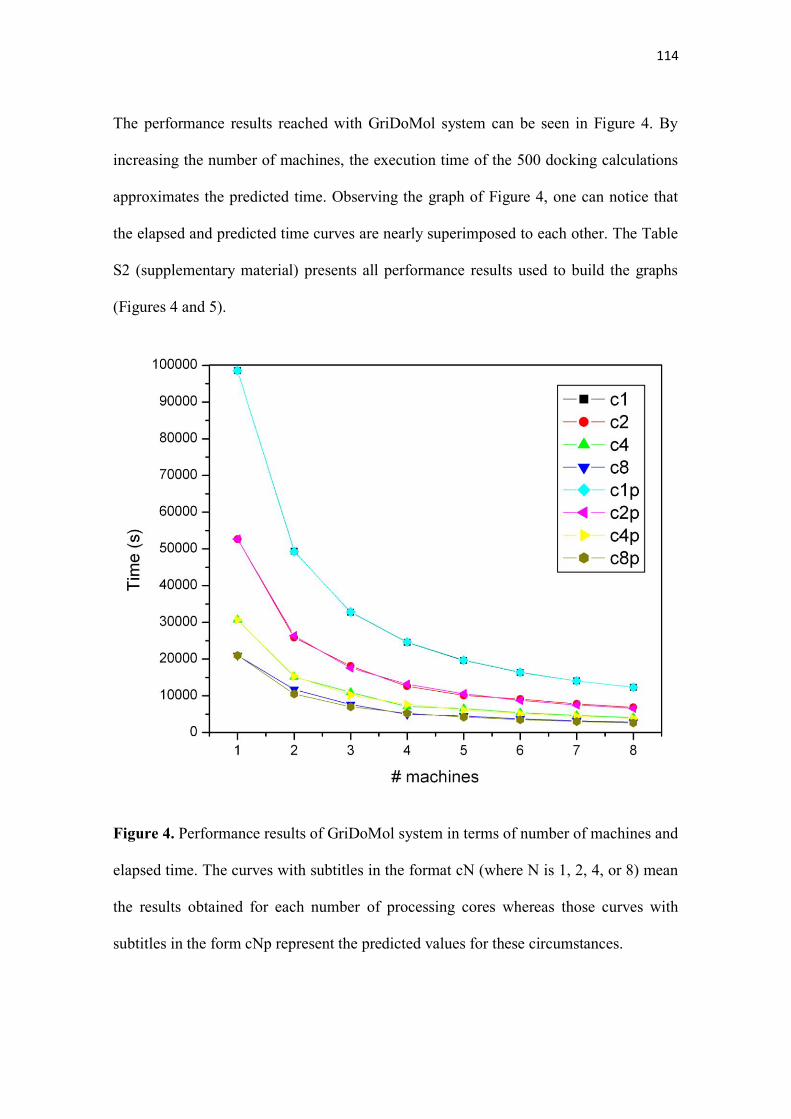
114
The performance results reached with GriDoMol system can be seen in Figure 4. By
increasing the number of machines, the execution time of the 500 docking calculations
approximates the predicted time. Observing the graph of Figure 4, one can notice that
the elapsed and predicted time curves are nearly superimposed to each other. The Table
S2 (supplementary material) presents all performance results used to build the graphs
(Figures 4 and 5).
Figure 4. Performance results of GriDoMol system in terms of number of machines and
elapsed time. The curves with subtitles in the format cN (where N is 1, 2, 4, or 8) mean
the results obtained for each number of processing cores whereas those curves with
subtitles in the form cNp represent the predicted values for these circumstances.
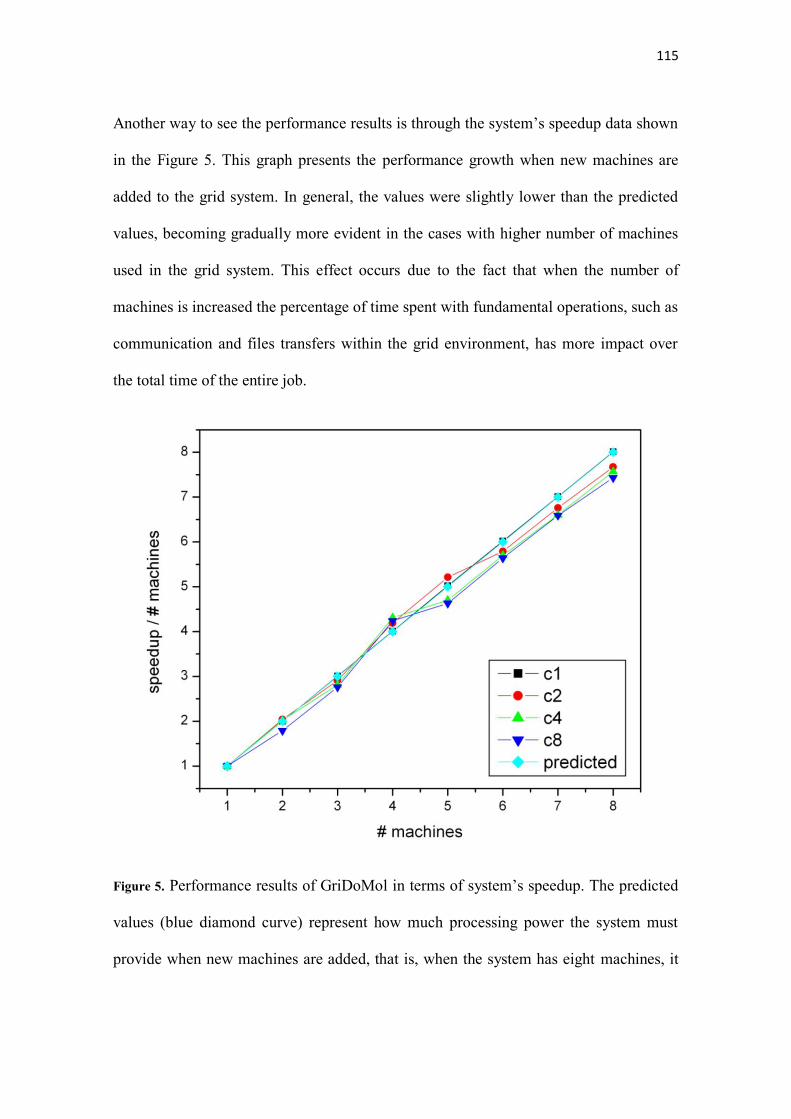
115
Another way to see the performance results is through the system’s speedup data shown
in the Figure 5. This graph presents the performance growth when new machines are
added to the grid system. In general, the values were slightly lower than the predicted
values, becoming gradually more evident in the cases with higher number of machines
used in the grid system. This effect occurs due to the fact that when the number of
machines is increased the percentage of time spent with fundamental operations, such as
communication and files transfers within the grid environment, has more impact over
the total time of the entire job.
Figure 5. Performance results of GriDoMol in terms of system’s speedup. The predicted
values (blue diamond curve) represent how much processing power the system must
provide when new machines are added, that is, when the system has eight machines, it

116
should perform a given docking calculation eight times faster than the same simulation
done by only one machine.
In the tests carried out on this work, the performance speedup of the individual docking
calculation using eight processing cores ranged from 4.02 to 7.38 times faster in
comparison to the calculation using a single core. Based on what has been observed in
the original study of AutoDock Vina [14], the average value of speedup with eight
processing cores reported in this reference was 7.25 times, lower than expected
performance of 8 times for this quantity of cores. This variation of performance at
processing cores level between the GriDoMol tests and the original study of AutoDock
Vina may have occurred due to differences in machine configuration (software and
hardware) used in each of these analysis. It is worth mentioning that the purpose of this
study was not to optimize the internal parallelization performance of AutoDock Vina
program, but just to measure the performance gain through the use of a grid computing
environment.
In order to analyze how much the file transfer phase impacts the total time of the whole
job, some jobs (group of 500 re-docking calculations) were re-executed without the file
transfer step, which means that all the files needed for the tests were previously made
available on each machine. The comparison of the total time of calculations in the cases
“with” and “without” the file transfer phase gives a measurement of the time spent on
this step (file transfer). The analysis of the data obtained with this new test revealed that
the time spent on file transfer phase was 9.39% of the total time, when one machine was
used in the computational grid. Additionally, when it was used five machines in the
grid, the contribution of the file transfer step was measured as 3.23% of the time spent
in the overall job. We believe that this happened because more machines were

117
performing the docking calculations, consequently; there was a higher chance that,
fortuitously, some of these file transfers may be occurring simultaneously among the
machines, reducing the total time spent on the job.
Discussion
In general, examining all GriDoMol’s tests and considering the performance of the
computational grid, it was noticed that when using more machines, rather than more
processing cores, the docking calculation proved to be more efficient, taking a small
amount of time. For instance, the use of eight machines and one core for each machine
(12,312 seconds) is faster than the inverse, i.e., one machine and the eight cores for it
(20,977 seconds). When using both strategies to distribute the docking calculations, grid
computing and multicore processing, and considering the infrastructure used in this
work (eight machines containing eight cores each), GriDoMol system provided a
reduction of the overall calculation time by up to 97% of the time spent in a non-
parallelized calculation (one machine with one core running). So, the use of these
distributed computing strategies (grid computing and multiprocessing) brought a
performance gain up to 34.91 times within GriDoMol system. Besides, due to the
scalability of such computational grid systems, if more machines are added to the grid,
even a higher speed up could be obtained.
Conclusion
On this present work, we have found the factors that cause speed up and the limitations
which impact the data processing, file transfer and communication phases. The
simultaneous occurrence of these phases on a distributed system, even if these
occurrences happen on different phases (e.g. a given machine is in file transfer phase

118
while the other is already in data processing phase), harshly reduces the required time to
accomplish a given number of tasks if compared to these phases being done sequentially
(one machine), thus reducing the overall time required to perform the set of tasks.
On the other hand, the idleness of data processing, file transfer and communication
phases represents some of the limitations of this type of distributed system. The idleness
at this data processing phase happens when there are more machines available than
tasks to be performed. For example, when performing the final 3 tasks on a
computational grid with 10 machines, 7 machines will be idle waiting for the remaining
machines to finish these last tasks. There is also idleness on the file transfer and
communication phases on a computational grid if compared to a standalone execution.
Even with these limitations mentioned above, speed up improvement achieved by using
both parallelism strategy, grid level (eight machines) and core level (eight cores by
machine), was 34.91 times faster than using only one machine and one core. Thus, the
time required to perform 500 tasks of molecular docking was reduced by up to 97% of
the time required when compared to not using any parallelism strategy or distributed
computing.
Therefore, we conclude that the development of the software GriDoMol, which is an
integrated system that allows the use of distributed computing resources offered by the
computational grid environment OurGrid, in combination with the program AutoDock
Vina to perform molecular docking, achieved its primary goal of increasing the
computational performance, allowing the execution of a large set of individual tasks
("BoT") in a small fraction of the time it would take to perform the same set of tasks
sequentially on a single computer.

119
ASSOCIATED CONTENT
Supporting Information. Table S1 with the complete list of complexes used for the
performance tests and the Table S2 containing the detailed performance results. This
material is available free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author
*E-mail: [email protected]
ACKNOWLEDGMENT
The author Luiz Felipe G. R. Ferreira would like to thank the scholarship support from
Fundação de Amparo à Ciência e Tecnologia de Pernambuco - FACEPE. Authors also
would like to thank CNPq for financial support.
REFERENCES
(1) Geldenhuys, W. J.; Gaasch, K. E.; Watson, M.; Allen, D. D.; Van der Schyf, C. J.
Optimizing the use of open-source software applications in drug discovery, Drug
Discov. Today, 2006 11, 127-132.
(2) McGee, P. Modeling success with in silico tools, Drug Discov. Develop., 2005, 8,
24–28.
(3) Araujo, E.; Cirne, W.; Wagner, G.; Oliveira, N.; Souza, E. P.; Galvao, C. O.;
Martins, E. S. The SegHidro experience: Using the grid to empower a hydro-
meteorological scientific network, Ieee Computer Soc., Los Alamitos, 2005.

120
(4) Mattos, G.; Formiga, A. D.; Lins, R. D.; Martins, F. M. J. BigBatch: A Document
Processing Platform for Clusters and Grids, Assoc. Computing Machinery, New York,
2008.
(5) Pappalardo, F.; Halling-Brown, M. D.; Rapin, N.; Zhang, P.; Alemani, D.; Emerson,
A.; Paci, P.; Duroux, P.; Pennisi, M.; Palladini, A.; Miotto, O.; Churchill, D.; Rossi, E.;
Shepherd, A. J.; Moss, D. S.; Castiglione, F.; Bernaschi, M.; Lefranc, M. P.; Brunak, S.;
Motta, S.; Lollini, P. L.; Basford, K. E.; Brusic, V. ImmunoGrid, an integrative
environment for large-scale simulation of the immune system for vaccine discovery,
design and optimization, Brief. Bioinform., 2009, 10, 330-340.
(6) Giupponi, G.; Harvey, M. J.; De Fabritiis, G. The impact of accelerator processors
for high-throughput molecular modeling and simulation, Drug Discov. Today, 2008, 13,
1052-1058.
(7) Woods, C. J., Ng, M. H.; Johnston, S.; Murdock, S. E.; Wu, B.; Tai, K.; Fangohr,
H.; Jeffreys, P.; Cox, S.; Frey, J. G.; Sansom, M. S. P.; Essex, J. W. Grid computing and
biomolecular simulation, Philos. Trans. R. Soc. A-Math. Phys. Eng. Sci., 2005, 363,
2017-2035.
(8) Buyya, R.; Branson, K.; Giddy, J.; Abramson, D. The virtual laboratory: A toolset
for utilizing the world-wide grid to design drugs, Ieee Computer Soc, Los Alamitos,
2002.
(9) Zhang, S. X.; Kumar, K.; Jiang, X. H.; Wallqvist, A.; Reifman J. DOVIS: an
implementation for high-throughput virtual screening using AutoDock, BMC
Bioinformatics, 2008, 9.

121
(10) De Vries, S. J.; van Dijk, M.; Bonvin, A. The HADDOCK web server for data-
driven biomolecular docking, Nat. Protoc., 2010, 5, 883-897.
(11) Abreu, R. M. V.; Froufe, H. J. C.; Queiroz, M.; Ferreira, I. MOLA: a bootable,
self-configuring system for virtual screening using AutoDock4/Vina on computer
clusters, J. Cheminformatics, 2010, 2.
(12) Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D.
S.; Olson, A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective
Receptor Flexibility, J. Comput. Chem., 2009, 30, 2785-2791.
(13) e-NMR Web Portal. http://www.enmr.eu/webportal/ (31/01/13)
(14) Trott, O.; Olson, A. J. Software News and Update AutoDock Vina: Improving the
Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization,
and Multithreading, J. Comput. Chem., 2010, 31, 455-461.
(15) Andrade, N.; Cirne, W.; Brasileiro, F.; Roisenberg, P. OurGrid: An approach to
easily assemble grids with equitable resource sharing, in: D. Feitelson, L. Rudolph, U.
Schwiegelshohn (Eds.) Job Scheduling Strategies for Parallel Processing, Springer-
Verlag Berlin, Berlin, 2003, 61-86.
(16) Ignite Realtime: Openfire Server. http://www. igniterealtime.org/projects/openfire/
(10/12/12).
(17) Qt framework. http://qt.digia.com/ (10/12/12).

122
(18) Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of
eight docking tools for docking and virtual screening accuracy, Proteins, 2004, 57, 225-
242.


















