

BPF
201
cGMP cGMP “Current Good Manufacturing Practices” BPF BPF (“Boas Práticas de Fabricação”) Valéria dos S. Cozzolino Yugue HSV Consultores Associados Ltda. Junho/2001
-
Upload
rodrigo-noguerol-correa -
Category
Documents
-
view
3 -
download
0
description
Boas práticas de fbricação
Transcript of BPF

cGMPcGMP“Current Good Manufacturing
Practices”BPFBPF (“Boas Práticas de
Fabricação”)Valéria dos S. Cozzolino Yugue
HSV Consultores Associados Ltda.Junho/2001

cGMPcGMP
• Todos precisam conhecer• Não somente por ser
mandatório• Mas por ser um “bom
negócio”!

cGMPcGMP
•Certeza de um produto seguro e efetivo

Vamos ver como vocês se sentem em relação
ao cGMP...

Exercício IExercício IPesquisa(5 minutos)

PontuaçãoPontuação
Sempre 5
Frequentemente 4
Algumas vezes 3
Raramente 2
Nunca 1

O que significa?O que significa?
45 - 50 Você está indomuito bem!
39 - 44 Você está indorazoavelmente bem
mas deveriatrabalhar para
melhorar.33 - 38 Você precisa
colocar um esforçoreal na melhoria de
seu GMP.Menos que 32 Você deve fazer um
esforço paramelhorar seu GMP.

O que você espera de O que você espera de um treinamento de um treinamento de
cGMP ?cGMP ?

O que você pensa O que você pensa quando o assunto é quando o assunto é
cGMP ?cGMP ?O que é cGMP ?O que é cGMP ?

Objetivos do CursoObjetivos do Curso
• Focar o estudo e conhecimento do: – Regulamento Técnico:Regulamento Técnico: Boas Práticas de
Fabricação para os estabelecimentos produtores de medicamentos (Agência Nacional de Vigilância Sanitária - MS)
• Esclarecer dúvidas em relação ao cGMP• Homogeneizar o grau de conhecimento
no assunto dentro do grupo

MetodologiaMetodologia
• Percorrer a norma, analisando cada item criticamente
• O que temos?• O que falta completar?• O que não temos?• O que é preciso para
melhorarmos?

Quem é responsável pela Quem é responsável pela aderência às normas de aderência às normas de
cGMP?cGMP?• Gerências têm
responsabilidade• CQ/GQ têm
responsabilidade• Cada colaborador
tem responsabilidade (incluindo você!)

Gerenciamento da Qualidade Gerenciamento da Qualidade na Indústria Farmacêuticana Indústria Farmacêutica
O Gerenciamento da Qualidade é o aspecto da função de gerenciamento
que determina e implementa a “Política da Qualidade”, ou seja, as
intenções e direções globais relativas à qualidade, formalmente
expressa e autorizada pela administração superior da empresa.

Os elementos básicos do Os elementos básicos do Gerenciamento da Qualidade na Gerenciamento da Qualidade na Indústria de Medicamentos sãoIndústria de Medicamentos são::
• Uma infra-estrutura apropriada ou "sistema de qualidade", englobando a estrutura organizacional, os procedimentos, os processos e os recursos;
• Ações sistemáticas e precisas para assegurar que determinado produto (ou serviço) satisfaça as exigências quanto à sua qualidade. A totalidade dessas ações é chamada "Garantia da Qualidade".

• Os conceitos de Garantia da Qualidade, de GMP e de Controle de Qualidade são aspectos inter-relacionados do gerenciamento da qualidade.
• Suas relações são de fundamental importância para a produção e para o controle de medicamentos.
GQCQ

Garantia da QualidadeGarantia da Qualidade
Dentro de uma organização, a Garantia da Qualidade serve como ferramenta de gerenciamento. Em situações contratuais, a Garantia da Qualidade serve também para gerar confiança no fornecedor.

Um sistema apropriado da Um sistema apropriado da Garantia Garantia da Qualidadeda Qualidade, aplicado à fabricação , aplicado à fabricação de medicamentos, deve assegurar de medicamentos, deve assegurar
que: que: • Os medicamentos sejam planificados e
desenvolvidos considerando a necessidade do cumprimento das cGMPs e outros aspectos associados às Boas Práticas de Laboratório (BPL) e às Boas Práticas Clínicas (BPC);
• As operações de produção e controle sejam claramente especificadas por escrito e as exigências de cGMP cumpridas;

Um sistema apropriado da Garantia Um sistema apropriado da Garantia da Qualidade, aplicado à fabricação da Qualidade, aplicado à fabricação de medicamentos, deve assegurar de medicamentos, deve assegurar
que: (cont.)que: (cont.)• As responsabilidades gerenciais estejam
claramente especificadas na descrição dos procedimentos;
• Sejam tomadas providências quanto à fabricação, ao suprimento e à utilização correta das matérias-primas e materiais de embalagem;
• Todos os controles necessários sejam realizados nas matérias-primas, produtos intermediários, produtos a granel e produto acabado, como realizar outros controles necessários durante o processo, além das calibrações e das validações;

• O produto acabado seja corretamente processado e conferido, segundo procedimentos definidos;
• Os medicamentos não sejam vendidos ou fornecidos antes que o pessoal autorizado confirme, que cada um dos lotes tenha sido fabricado e controlado de acordo com os requisitos do registro e os regulamentos relevantes a produção, controle e liberação;
Um sistema apropriado da Garantia Um sistema apropriado da Garantia da Qualidade, aplicado à fabricação da Qualidade, aplicado à fabricação de medicamentos, deve assegurar de medicamentos, deve assegurar
que: (cont.)que: (cont.)

• Garantir, que os medicamentos sejam armazenados, distribuídos e subseqüentemente manuseados, de forma que a qualidade dos mesmos seja mantida por todo o prazo de validade;
• Procedimento de auto-inspeção e/ou auditoria interna de qualidade que avalie regularmente a efetividade e a aplicação do sistema de Garantia da Qualidade.
Um sistema apropriado da Garantia Um sistema apropriado da Garantia da Qualidade, aplicado à fabricação da Qualidade, aplicado à fabricação de medicamentos, deve assegurar de medicamentos, deve assegurar
que: (cont.)que: (cont.)

Boas Práticas de Boas Práticas de FabricaçãoFabricação

Boas Práticas de Boas Práticas de FabricaçãoFabricação
• Processos de fabricação definidos e sistematicamente revisados. Devem ser capazes de fabricar medicamentos consistentes, atendendo às respectivas especificações;
• Etapas críticas e quaisquer modificações significativas nos processos de produção e controle de qualidade devem ser sistematicamente validados;

Boas Práticas de Boas Práticas de Fabricação (cont.)Fabricação (cont.)
• As área de produção devem ser providas de toda a infra-estrutura necessária: – Pessoal qualificado e devidamente treinado; – Espaço e instalações adequadas; – Equipamento e serviços adequados; – Materiais, recipientes e rótulos corretos; – Procedimentos e instruções aprovadas; – Armazenamento e transporte adequados;– Laboratórios, equipamentos e pessoal adequado, para
controle em processo sob a responsabilidade da gerência de produção;

Boas Práticas de Boas Práticas de Fabricação (cont.)Fabricação (cont.)
• Instruções e procedimentos escritos em linguagem clara e inequívoca e aplicáveis de forma específica às instalações disponíveis;
• Operadores devem ser treinados para desempenharem corretamente os procedimentos;
• Devem ser feitos registros durante a fabricação, de modo a demonstrar que todas as providências exigidas pelos procedimentos e instruções tenham sido tomadas e que a quantidade e a qualidade do produto estejam em conformidade com o programado. Quaisquer desvios significativos devem ser registrados e investigados;

Boas Práticas de Boas Práticas de Fabricação (cont.)Fabricação (cont.)
• Registros que possibilitem um rastreamento do lote;
• Armazenamento adequado e a distribuição dos produtos devem minimizar qualquer risco à sua qualidade;
• Sistema capaz de recolher qualquer lote, após sua venda ou fornecimento;
• Reclamações sobre produtos comercializados e as causas dos desvios de qualidade investigadas.

Controle de Controle de QualidadeQualidade

Controle de Controle de QualidadeQualidade
• O controle de qualidade é a parte do cGMP referente à amostragem, às especificações, aos ensaios, aos procedimentos de organização, à documentação e aos procedimentos de liberação que devem garantir que os ensaios necessários e relevantes sejam executados e que os materiais não sejam liberados para uso (nem os produtos liberados para venda ou fornecimento) até que a qualidade dos mesmos seja julgada como satisfatória.

• Disponibilidade de instalações adequadas, pessoal treinado e procedimentos operativos aprovados para que se possa realizar as amostragens, a inspeção e os ensaios das matérias-primas, dos materiais de embalagem, dos produtos intermediários, dos produtos a granel e dos produtos acabados e, quando necessário para o monitoramento das condições ambientais das áreas limpas.
• Todas as metodologias dos ensaios de controle de qualidade devem ser validadas.
Controle de Controle de Qualidade Qualidade (cont.)(cont.)

• Os registros devem ser feitos , de modo a demonstrar que todos os procedimentos de amostragem, inspeções e ensaios requeridos tenham sido realmente executados e que quaisquer desvios tenham sido totalmente registrados, investigados e corrigidos.
• Os produtos acabados devem conter princípios ativos, que atendam à composição quantitativa e qualitativa do registro do produto e devem apresentar a pureza exigida, estar embalados em material adequado e corretamente rotulados.
Controle de Controle de Qualidade Qualidade (cont.)(cont.)

• Deve ser feito o registro dos resultados obtidos da inspeção e os ensaios de controle dos materiais, dos produtos intermediários, a granel e acabados, quanto ao atendimento das especificações. A avaliação dos lotes de produtos deve incluir a revisão e a avaliação da documentação de produção, bem como a avaliação dos desvios aos procedimentos específicos .
Controle de Controle de Qualidade Qualidade (cont.)(cont.)

• Nenhum lote de produto pode ser liberado para venda ou fornecimento antes de ser aprovado, em conformidade com as exigências previstas no registro oficial.
• Devem ser retiradas amostras suficientes das matérias-primas, do material de embalagem e do produto acabado, a fim de que possam ser feitos, se necessário, exames futuros do produto; as amostras de produto acabado retidas devem ser mantidas em suas embalagens finais, a menos que as mesmas sejam excepcionalmente grandes.
Controle de Controle de Qualidade Qualidade (cont.)(cont.)

Sanitização e HigieneSanitização e Higiene

Sanitização e HigieneSanitização e Higiene• Nosso produto - Medicamentos• Exige alto grau de sanitização e higiene• Deve abranger:
– Pessoal– Instalações– Equipamentos– Materiais– Produtos de Limpeza e desinfecção

ValidaçãoValidação

ValidaçãoValidação• Processos críticos devem ser validados,
prospectiva e/ou retrospectivamente.• Novos processos, quando introduzidos,
deve-se demonstrar por validação, a adequação do novo método.
• O processo definido, deve mostrar-se capaz de dar origem a produtos consistentes, dentro de padrões de qualidade exigidos.

ValidaçãoValidação• Qualquer modificação significativa
deve ser validada.Equipamento
Área de fabricação
Processo
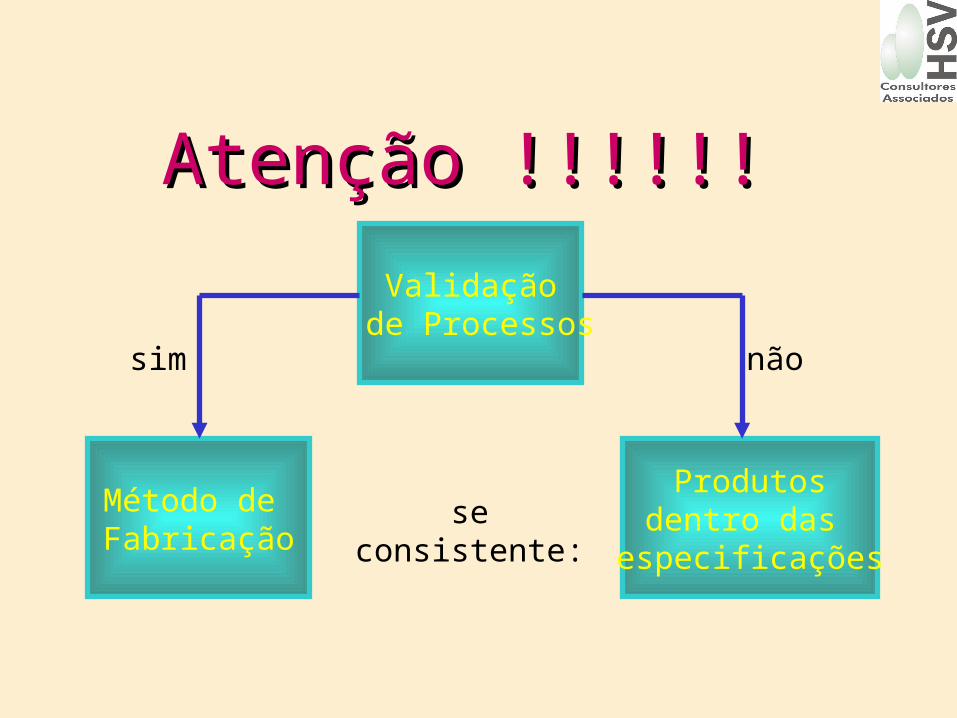
Atenção !!!!!!Atenção !!!!!!
Validação de Processos
sim não
se consistente:Produtos
dentro das especificações
Método de Fabricação

ResponsabilidadesResponsabilidades
Validação é um
trabalho de equipe!

Desenvolvimento
Garantiade
Qualidade
Produção
Engenharia/Manutenção
qualificação e certificação
informação sobre
capabilidade do processo
Aprovação de protocolos e condutas de
validação
operação e manutenção
da planta
ReclamaçõesReclamações

ReclamaçõesReclamações• Todas as reclamações e demais
informações referentes a produtos com desvios potenciais de qualidade devem ser cuidadosamente investigadas, seguindo procedimentos escritos.

Recolhimento de Recolhimento de ProdutosProdutos

Recolhimento de Recolhimento de ProdutosProdutos
• Deve haver um sistema que retire imediata e efetivamente do mercado os produtos que apresentarem desvios de qualidade ou que estejam sob suspeita.

ContratosContratosFabricação e Análise

Contratos de fabricação e Contratos de fabricação e análise de Controle de análise de Controle de
QualidadeQualidade
• O contrato de fabricação e/ou análise de controle de qualidade deve ser mutuamente acordado e controlado entre as partes, de modo a evitar-se equívocos que possam resultar em processo, produto ou análise de qualidade insatisfatórias.

Contratos de fabricação e Contratos de fabricação e análise de Controle de análise de Controle de
QualidadeQualidade• Deve estabelecer os procedimentos de
fabricação e/ou análise.• Deve permitir que o contratante faça
auditorias às instalações do contratado.• Deve-se assegurar que as normas de
cGMP sejam cumpridas.

Auto-inspeção e Auto-inspeção e Auditoria da QualidadeAuditoria da Qualidade

Auto-inspeção e Auditoria da Auto-inspeção e Auditoria da QualidadeQualidade
• O objetivo da auto- inspeção é avaliar o cumprimento das cGMPs em todos os aspectos da produção e do controle da qualidade.
• Deve ser capaz de detectar todas as deficiências na implementação das cGMPs.

Auto-inspeção e Auditoria da Auto-inspeção e Auditoria da QualidadeQualidade
• Quando deve ser realizada?– Rotineiramente– Recolhimento de produtos– Rejeições repetidas– Previamente a uma inspeção por
autoridades sanitárias – A frequência deve estar relacionada ao
tipo de produto fabricado e às necessidades da empresa

Auto-inspeção e Auditoria da Auto-inspeção e Auditoria da QualidadeQualidade
• Quem deve realizar?– Pessoal que possa avaliar com
objetividade a implementação das cGMPs.

Auto-inspeção e Auditoria da Auto-inspeção e Auditoria da QualidadeQualidade
• Todas as recomendações sobre medidas corretivas devem ser implementadas.
• Os procedimentos para a realização da auto-inspeção devem ser documentados e possuir um programa efetivo de acompanhamento.

Ítens mínimos para Auto-Ítens mínimos para Auto-inspeçãoinspeção
• Pessoal• Instalações• Manutenção de
prédios e equipamentos
• Armazenamento de matéria prima e de produtos acabados

Ítens mínimos para auto-inspeção Ítens mínimos para auto-inspeção (cont.)(cont.)
• Equipamentos• Produção e controles
em processo• Controle de
qualidade• Documentação• Sanitização e higiene• Programas de
validação e revalidação

• Calibração de instrumentos ou de sistemas de medidas
• Procedimentos de recolhimento do mercado
Ítens mínimos para auto-inspeção Ítens mínimos para auto-inspeção (cont.)(cont.)

• Gerenciamento de reclamações
• Controle de rótulos• Resultados de
auto-inspeções anteriores e quaisquer passos corretivos tomados
Ítens mínimos para auto-inspeção Ítens mínimos para auto-inspeção (cont.)(cont.)

Relatório de auto-inspeçãoRelatório de auto-inspeção
• Resultados• Avaliações e
conclusões• Ações
corretivas recomendadas

Ações de acompanhamentoAções de acompanhamento
• Verificação do cumprimento das ações corretivas - relatório específico

Auditoria da QualidadeAuditoria da Qualidade
• Complementação da auto-inspeção• Avaliação de todo ou parte do
Sistema de Qualidade - objetivo de aperfeiçoamento
• Em geral, é realizada por especialistas externos
• Devem ser extendidas aos fornecedores e contratados

Auditoria de FornecedoresAuditoria de Fornecedores
• Responsabilidade - CQ ou GQ e demais departamentos envolvidos na produção
• Avaliação e aprovação para inclusão na lista de fornecedores da empresa

PessoalPessoal

PessoalPessoal
• O estabelecimento e a manutenção de um sistema satisfatório de Garantia da Qualidade e a correta fabricação e controle de medicamentos dependem das pessoas que os realizam.

Educação e TreinamentoEducação e Treinamento
“Qualidade começa e termina com educação”
K. Ishikawa
“Qualidade começa e termina com educação”
K. Ishikawa

PessoalPessoal
Todas as responsabilidades individuais devem estabelecer-se em procedimentos escritos e ser claramente compreendidas pelo
indivíduos envolvidos. Todo pessoal deve conhecer os
princípios das cGMPs que os afetam.

PessoalPessoal
Todo o pessoal deve ser motivado a apoiar a empresa e a manutenção de altos padrões de qualidade .

PessoalPessoalTreinamentoTreinamento
Programa escrito e definido de treinamento às pessoas envolvidas
nas áreas de produção ou nos laboratórios de controle de
qualidade (incluindo o pessoal técnico, de manutenção e de
limpeza), e a todo pessoal cujas atividades possam afetar a
qualidade do produto.

PessoalPessoalTreinamentoTreinamento
Visitantes e pessoas não treinadas, não devem ser levados para dentro das
áreas de produção. Se isso for inevitável, essas pessoas devem ser antecipadamente informadas sobre a
higiene pessoal e o uso de vestimentas protetoras e devem ser
acompanhadas por pessoal autorizado.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Exame de saúde no momento de admissão e exames médicos periódicos, de acordo com
procedimentos estabelecidos.• As pessoas que fazem inspeção
visual devem fazer exames oftalmológicos periodicamente.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Treinamento nas práticas de higiene pessoal. Todas as pessoas envolvidas nos processos de fabricação devem cumprir com as normas de higiene; particularmente, devem ser instruídas a lavarem suas mãos antes de entrarem nas áreas de produção.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Qualquer pessoa aparentemente enferma ou que tiver lesões expostas que possam afetar de forma adversa a qualidade dos produtos, não pode manusear matérias-primas, materiais de embalagem, produtos intermediários e a granel ou produtos acabados até que sua condição de saúde não represente risco ao produto.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Todos os empregados devem ser instruídos e incentivados a reportar a seus supervisores imediatos quaisquer condições (relativas à área de produção, ao equipamento ou ao pessoal) que considerem que possam afetar adversamente os produtos.

PessoalPessoalHigiene PessoalHigiene Pessoal
Deve ser evitado o contato direto entre as mãos do operador e as matérias-primas, os materiais de
embalagem primários, os produtos intermediários e a granel.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Para que seja assegurada a proteção do produto contra contaminação, o pessoal deve vestir roupas limpas e apropriadas à área de produção, essas roupas devem incluir capuzes apropriados.
• Os uniformes, se forem reutilizáveis, devem ser guardados em ambientes fechados, até que sejam lavados e, se necessários desinfetados ou esterilizados.

PessoalPessoalHigiene PessoalHigiene Pessoal
• Não permitido fumar, comer, beber, mascar ou manter plantas, alimentos, bebidas, fumo e medicamentos pessoais nas áreas de produção, do laboratório de controle de qualidade e de armazenamento, ou em quaisquer outras áreas em que tais ações possam influir adversamente na qualidade do produto.

Filme:Filme:
“Quem roubou o meu queijo?

InstalaçõesInstalações
• As instalações devem ser localizadas, projetadas, construídas, adaptadas e mantidas de forma que sejam adequadas às operações a serem executadas.
• Seu projeto deve minimizar o risco de erros e possibilitar a limpeza e manutenção, de modo a evitar a contaminação cruzada, o acúmulo de poeira e sujeira ou qualquer efeito adverso que possa afetar a qualidade dos produtos.

InstalaçõesInstalações(Áreas auxiliares)(Áreas auxiliares)
• Os vestiários, lavatórios e os sanitários devem ser de fácil acesso e apropriados para o número de usuários.
• Os sanitários não devem ter comunicação direta com as áreas de produção e armazenamento.

InstalaçõesInstalações(Áreas auxiliares)(Áreas auxiliares)
• As áreas de manutenção devem situar-se em locais separados das áreas de produção.
• Se as ferramentas e peças de reposição, são mantidas nas áreas de produção, as mesmas devem se mantidas em salas ou armários reservados para este fim.

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
• Estoque ordenado e adequado de várias categorias de materiais e produtos: • matérias-primas• materiais de embalagem• materiais intermediários• granel• produtos acabados
• Identificação de sua condição: quarentena, liberado, reprovado, devolvido ou recolhido.

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
• As áreas de recebimento devem ser projetadas e equipadas de forma a permitir que os recipientes de materiais recebidos sejam limpos antes de serem estocados Gerenciamento de reclamações
• Controle de rótulos• Resultados de auto-inspeções anteriores e
quaisquer passos corretivos tomados

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
Os produtos em quarentena devem estar em área restrita e separadas na área de armazenamento, essas
áreas devem ser claramente demarcadas e o acesso às mesmas
somente pode ser efetuado por pessoas autorizadas.

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
• Deve haver uma área separada para a coleta de amostras das matérias-primas.
• Se a amostragem for feita na área de armazenamento, ela deve ser realizada em um ambiente específico para essa finalidade, de forma que não haja possibilidade de contaminação microbiológica e/ou contaminação cruzada.

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
• O armazenamento de materiais ou produtos devolvidos, reprovados ou recolhidos deve efetuar-se em área separada e identificada.
• Os materiais altamente ativos, narcóticos, produtos perigosos, substâncias controladas, ou que apresentem riscos de incêndio ou explosão, devem ser estocados em áreas seguras e protegidas, devidamente segregados e identificados.

InstalaçõesInstalações(Áreas de armazenamento)(Áreas de armazenamento)
• Deve dar-se especial atenção ao armazenamento de materiais impressos.
• Estes devem ser guardados em local fechado, evitando misturas e desvios.
• O material impresso só pode ser manuseado por pessoal autorizado, seguindo procedimentos definidos e escritos.

InstalaçõesInstalações(Áreas de pesagem)(Áreas de pesagem)
• A pesagem das matérias-primas deve ser realizada em áreas separadas, projetadas para esse fim, com instalações de exaustão adequada, para controlar o pó formado.

InstalaçõesInstalações(Áreas de produção)(Áreas de produção)
• Devem existir instalações exclusivas e separadas para a produção de alguns medicamentos altamente sensibilizantes (por exemplo, penicilina e seus derivados) ou produtos biológicos, microorganismos vivos). A produção de determinados medicamentos, como alguns antibióticos, hormônios, substâncias citotóxicas e produtos altamente ativos, não deve realizar-se nas mesmas instalações.

InstalaçõesInstalações(Áreas de produção)(Áreas de produção)
• Nas áreas onde estiverem expostos ao ambiente as matérias-primas, os materiais de embalagem primários, os produtos intermediários ou a granel, as superfícies interiores (paredes, piso e teto) devem ser lisas e livres de rachaduras e de juntas abertas. Não devem liberar partículas e devem permitir uma limpeza fácil e efetiva.

InstalaçõesInstalações(Áreas de produção)(Áreas de produção)
• As tubulações, iluminação, pontos de ventilação e outros serviços devem ser projetados e instalados de modo a evitar a criação de áreas de difícil limpeza. Sempre que possível sua manutenção deve ser efetuada externamente das áreas de produção.

InstalaçõesInstalações(Áreas de produção)(Áreas de produção)
• Devem possuir sistema de ventilação efetivo, com unidades de controle de ar (incluindo o controle de temperatura e, quando necessário, de umidade e filtração) apropriados aos produtos nela manipulados, às operações realizadas e às condições do ambiente.

InstalaçõesInstalações(Áreas de produção)(Áreas de produção)
• Os laboratórios de controle de qualidade devem ser totalmente separados das áreas de produção. As áreas onde forem realizados os ensaios microbiológicos, devem ser independentes e adequadamente separadas.
• Deve haver espaço suficiente e adequado para o armazenamento de amostras de referência e das referências ou padrões e da documentação do registros dos lotes.
• Deve possuir sistema de ventilação e prevenir a formação de vapores nocivos.

MATERIAL ERRADO
FERRUGEM, ÓLEO
VIDRO, METAL
CABELO, FIAPOS
CONTAMINAÇÃO GERALCONTAMINAÇÃO GERAL

EquipamentosEquipamentos
• Todas as tubulações e os instrumentos utilizados devem ser devidamente identificados.
• As balanças e outros equipamentos de medida disponíveis nas áreas de produção e de controle de qualidade, devem ter a capacidade e a precisão requerida e devem ser periodicamente calibrados.

EquipamentosEquipamentos
• Os equipamentos utilizados na produção devem ser projetados, dispostos e mantidos de forma a servirem ao uso ao qual tenham sido designados.
• Os equipamentos utilizados na produção devem ser projetados de forma que possam ser facilmente limpos.
• Os processos de limpeza e lavagem dos equipamento não devem constituir fonte de contaminação.

EquipamentosEquipamentos
• As partes destes equipamentos que entrarem em contato com o produto não devem ser reativas, aditivas ou absortivas de forma a influir na qualidade do produto.
• Todo equipamento avariado deve, se possível, ser retirado das áreas de produção e de controle de qualidade. Caso contrário, deve estar devidamente identificado de tal condição, usando-se etiquetas ou placas bem visíveis.

MateriaisMateriais
O objetivo principal de uma planta farmacêutica é produzir
medicamentos a partir da combinação de materiais (ativos, inativos e de embalagem), para
uso de pacientes e que devem, por este motivo, merecer atenção
especial.

MateriaisMateriais
• Devem ser postos em quarentena imediatamente após o recebimento ou produção, até que sejam liberados pelo controle de qualidade, para uso ou distribuição.

MateriaisMateriais
Todos os materiais e produtos devem ser armazenados sob condições apropriadas de acordo com os
procedimentos estabelecidos pelo fabricante, deve ser feita a
separação dos lotes e a rotatividade do estoque, obedecendo a regra:
primeiro que entra, primeiro que sai.

MateriaisMateriais(Matéria prima)(Matéria prima)
• As matérias primas devem estar adequadamente identificadas com, pelo menos:– Nome e código de referência interno do produto,
quando a empresa tenha estabelecido o sistema; – Número (s) do (s) lote atribuídos (s) pelo fornecedor e
o número dado pela empresa no momento do recebimento ;
– Situação interna do produto no armazenamento (exemplo: em quarentena, em análise, aprovado, reprovado, devolvido, recolhido );
– Data de fabricação, o prazo de validade e a data de reanálise .

MateriaisMateriais(Matéria prima)(Matéria prima)
• Devem ser utilizados procedimentos que garantam a identificação do conteúdo de
cada recipiente de matéria prima. Os recipientes a granel dos quais tenham sido
retiradas amostras, devem ser identificados.
• Somente as matérias primas liberadas pelo departamento de controle de qualidade e
que estejam dentro dos respectivos prazos de validade devem ser utilizadas.

MateriaisMateriais(Matéria prima)(Matéria prima)
Os materiais fracionados para cada lote de produção devem ser mantidos juntos e visivelmente identificados

MateriaisMateriais(Materiais de embalagem)(Materiais de embalagem)
• Devem ser estocados em condições seguras, para que o acesso não autorizado seja evitado.
• Os materiais de embalagem somente devem ser distribuídos para o uso, por pessoas designadas para tal, segundo procedimentos aprovados.
• Cada lote de material impresso e de material de embalagem deve receber um número específico de referência ou marca de identificação.
• Os materiais impressos, considerados antigos ou obsoletos devem ser destruídos e esse procedimento deve ser registrado.

MateriaisMateriais(Produtos intermediários e a (Produtos intermediários e a
granel)granel)Os produtos
intermediários e os produtos a granel adquiridos, devem ser manipulados no recebimento como se fossem matérias primas.

MateriaisMateriais(Produtos acabados)(Produtos acabados)
Os produtos acabados devem ser mantidos em quarentena até que sejam
finalmente liberados pelo controle de qualidade. Em seguida, devem ser
armazenados como estoque disponível, de acordo as condições estabelecidas.

MateriaisMateriais(Reprovados e devolvidos)(Reprovados e devolvidos)
• Armazenados separadamente, em áreas restritas. Podem ser devolvidos, reprocessados ou destruídos.
• O reprocessamento de produtos deve ocorrer somente em caráter excepcional. Somente será permitida se a qualidade do produto acabado não for afetada, se as especificações forem atendidas e se a operação for realizada de acordo com procedimentos autorizados e definidos após a avaliação dos riscos envolvidos.
• Qualquer lote reprocessado deve receber novo número de lote.

MateriaisMateriais(Reprovados e devolvidos)(Reprovados e devolvidos)
• A introdução da totalidade ou parte de lotes anteriores produzidos que atendam aos padrões de qualidade exigidos, a outro lote do mesmo produto, em determinado estágio da fabricação, deve ser previamente autorizada e ser realizada de acordo com procedimentos definidos, após a avaliação dos riscos envolvidos, inclusive qualquer possível efeito sobre o prazo de validade; o processo deve ser registrado.

MateriaisMateriais(Produtos recolhidos)(Produtos recolhidos)
• Os produtos recolhidos do mercado devem ser identificados e armazenados separadamente em área segura, até que seja tomada alguma decisão quanto aos seus destinos.

MateriaisMateriais(Produtos devolvidos)(Produtos devolvidos)
• Devem ser destruídos, a menos que a qualidade dos mesmos continue satisfatória. Somente podem ser considerados para revenda, reembalagem ou incorporados em outro granel de um lote subseqüente, após terem sido criticamente avaliados pelo departamento de controle da qualidade.
• Qualquer ação deve ser devidamente registrada.

MateriaisMateriais(Reagentes e meios de (Reagentes e meios de
cultura)cultura)• Devem ser registrados ao serem recebidos ou
preparados.• Os reagentes preparados em laboratório devem
ser elaborados de acordo com procedimentos escritos e apropriadamente rotulados. • O rótulo deve indicar concentração, data de
preparo, fator de padronização, prazo de validade, data de nova padronização e condições de armazenagem.
• Deve ser assinado e datado pela pessoa que preparou o reagente.

MateriaisMateriais(Padrões de referência)(Padrões de referência)
• Todos os padrões de referência devem ser guardados e utilizados de maneira que não tenham sua qualidade afetada.

MateriaisMateriais(Residuais)(Residuais)
• O material residual não deve ser acumulado. Ele deve ser coletado em recipientes adequados e colocado em pontos de coleta fora do prédio.
• Devem ser eliminados de forma segura e sanitária, a intervalos regulares e freqüentes.

MateriaisMateriais(Diversos)(Diversos)
• Não deve ser permitido que produtos rodenticidas, inseticidas, agentes fumigantes e materiais sanitizantes contaminem equipamentos, matérias primas, materiais de embalagem, materiais em processo ou produtos acabados.

Exercício IIExercício II
“cGMPgrama” individual

““cGMPgrama”cGMPgrama”Acredito e aplico Acredito e não aplico
Não acredito e aplico Não acredito e não aplico

DocumentaçãoDocumentação

Você acha que existe Você acha que existe muita documentação na muita documentação na
empresa?empresa?

• O cGMP requer completa documentação
• Todos os passos críticos de um processo devem ser controlados e registrados de forma detalhada.
• Provar que processos críticos fornecem resultados de encontro a atributos de qualidade requeridos através de um
trabalho de Validação.

““Não documentou ???Não documentou ???
Então nada aconteceu...Então nada aconteceu...

DocumentaçãoDocumentação
• Parte essencial do sistema de garantia da qualidade
• Define as especificações de todos os materiais e os métodos de fabricação e controle
• Informações necessárias para decisão de liberação ou não de determinado lote
• Possibilita rastreamento de qualquer lote sob suspeita de desvio de qualidade.

DocumentaçãoDocumentação(Rótulos)(Rótulos)
• Devem ser utilizados rótulos de cores diferentes, que indiquem a situação dos produtos no processo de produção (exemplo: em quarentena, aprovado, reprovado ou limpo).
• Os rótulos dos padrões de referência e documentos que os acompanhem devem indicar concentração, data de fabricação e prazo de validade, data em que tenha sido aberto e condições de armazenamento, quando necessário

DocumentaçãoDocumentação(Especificações e procedimentos de (Especificações e procedimentos de ensaio de Controle de Qualidade)ensaio de Controle de Qualidade)
• Os procedimentos de controle de qualidade devem ser validados, considerando as instalações e os equipamentos disponíveis.
• As especificações devem estar devidamente autorizadas e datadas.
• Os procedimentos dos ensaios devem ser aprovados e mantidos pelo Controle de Qualidade e estarem disponíveis.
• Revisões periódicas para que sejam obedecidas as novas edições da farmacopéia nacional ou outros compêndios oficiais.

DocumentaçãoDocumentação(Especificações para matérias (Especificações para matérias
primas e embalagem)primas e embalagem)• As especificações de matérias-primas e
materiais de embalagem primária ou materiais impressos devem possuir, no mínimo: • O nome e o código interno de referência • Referência se existir, da monografia
farmacopéica • Requisitos quantitativos e qualitativos
com os respectivos limites de aceitação.

DocumentaçãoDocumentação(Especificações para matérias (Especificações para matérias
primas e embalagem)primas e embalagem)• Os materiais de
embalagem devem atender às especificações, dando ênfase à compatibilidade dos mesmos com o produto farmacêutico que contêm.

DocumentaçãoDocumentação(Especificações para intermediários (Especificações para intermediários
e granel)e granel)• As especificações de produtos
intermediários e a granel devem estar disponíveis sempre que estes materiais forem adquiridos ou expedidos, ou se os dados sobre os produtos intermediários tiverem de ser utilizados na avaliação do produto final. As especificações devem ser compatíveis com às especificações relativas às matérias-primas ou aos produtos acabados.

DocumentaçãoDocumentação(Especificações para produtos (Especificações para produtos
acabados)acabados)• Devem incluir, no mínimo:
• Nome genérico do produto e marca ou denominação comercial, quando for o caso
• Nome(s) do(s) princípio(s) ativo(s) com suas respetivas DCB ou DCI;
• Composição do produto; • Identificação do lote • Forma farmacêutica e detalhes de embalagem• Orientação sobre amostragem e a realização de ensaio de
controle, e as referências utilizadas• Requisitos qualitativos ou quantitativos, com os respectivos
limites de aceitação• Condições e precauções a serem tomadas no armazenamento• Data de fabricação e prazo de validade.

DocumentaçãoDocumentação(Fórmula mestra)(Fórmula mestra)
• Deve existir uma fórmula mestra autorizada para cada produto e tamanho de lote a ser fabricado

DocumentaçãoDocumentação(Fórmula mestra)(Fórmula mestra)
• A fórmula padrão deve incluir: • Nome do produto com o código de referência relativo à sua
especificação; • Forma farmacêutica, concentração do produto e tamanho do
lote; • Lista de todas as matérias-primas a serem utilizadas (com seus
respectivos DCB e DCI); com a quantidade utilizada de cada uma, usando o nome genérico e referência que são exclusivos para cada material. Deve-se fazer menção a qualquer substância que possa desaparecer no decorrer do processo;
• Rendimento final esperado, com os limites aceitáveis, e dos rendimentos intermediários, quando for o caso;
• Indicação do local de processamento e do principal equipamento a ser utilizado;

DocumentaçãoDocumentação(Fórmula mestra)(Fórmula mestra)
• Os métodos (ou referência aos mesmos) a serem utilizados no preparo dos equipamentos, como limpeza (especialmente após mudança de produto), montagem, calibração e esterilização;
• Instruções detalhadas dos passos a serem dados no processamento, verificação dos materiais, pré- tratamentos, a seqüência da adição de materiais, tempos de mistura, temperaturas);
• Instruções relativas a quaisquer controles em processo com seus limites aceitáveis;
• Exigências relativas ao acondicionamento dos produtos, inclusive sobre o recipiente, sobre a rotulagem e sobre quaisquer condições especiais de armazenamento;
• Precauções especiais a serem observadas.

DocumentaçãoDocumentação(Instruções de acondicionamento (Instruções de acondicionamento
primário e secundário)primário e secundário)• Devem incluir os seguintes dados:
• Nome do produto; • Forma farmacêutica, concentração e via de aplicação, quando for o caso; • Dimensões, expressas em termos numéricos, peso ou volume do
produto contido no recipiente final; • Materiais de embalagem necessários para tamanho de lote padrão,
incluindo quantidades, tamanhos e tipos, com o código ou número de referência relativo as especificações de cada material
• Amostragem ou reprodução dos materiais utilizados no processo de embalagem, indicando o local onde tenham sido impressos ou gravados o número do lote e sua data de vencimento
• Garantia que não existam restos de material impresso nas linhas de embalagem
• Descrição das operações de embalagem• Controles em processo e instruções para amostragem e limites de
aceitação

DocumentaçãoDocumentação(Registros dos lotes de fabricação)(Registros dos lotes de fabricação)
• Registros devem se basear na fórmula mestra aprovada e em uso.
• Verificação se equipamentos e local de trabalho estão livres de produtos anteriores, e documentos e materiais necessários para o processo planejado. Limpeza de equipamentos.
• Todas as etapas devem ser registradas, contemplando o tempo inicial e o final de execução de cada operação, devidamente assinado e datada pelas pessoas responsáveis pela realização de cada etapa, corroborada pelo supervisor da área.

DocumentaçãoDocumentação(Registros de embalagem dos lotes)(Registros de embalagem dos lotes)
• Os registros dos lotes devem conter pelo menos as seguintes informações: • Nome do produto, número do lote produto a
granel e a quantidade a ser embalada, bem como o número de lote do produto acabado, a quantidade planejada de produto final que será obtida, a quantidade real obtida e a reconciliação
• Data(s) e a (a) hora (s) das operações de embalagem
• Nome da pessoa responsável pela operação de embalagem

DocumentaçãoDocumentação(Registros de embalagem dos lotes)(Registros de embalagem dos lotes)
• Iniciais dos operadores nas principais etapas; • Verificações feitas quanto à identificação e à
conformidade com as instruções para embalagem, incluindo os resultados dos controles em processo;
• Detalhes da operação de embalagem, incluindo referências aos equipamentos, às linhas de embalagem utilizadas e, quando necessário, a instruções relativas à manutenção dos produtos não embalados ou um registro dos produtos devolvidos que não tenham sido embalados, a ser devolvidos à área de armazenagem

DocumentaçãoDocumentação(Registros de embalagem dos lotes)(Registros de embalagem dos lotes)
• Sempre que possível, amostras dos materiais de embalagem impressos utilizados, inclusive daqueles que tragam o número de lote, a data de vencimento do prazo de validade e qualquer impressão adicional;
• Observações sobre problemas especiais, incluindo detalhes acerca de qualquer desvio das instruções fornecidas quanto ao processo de embalagem
• Quantidades e números de referência ou identificação de todos os materiais de embalagem impressos e dos produtos a granel entregues para serem embalados, utilizados, destruídos ou devolvidos ao estoque, e as quantidades de produto obtidas, a fim de que possa ser feita uma reconciliação adequada.

DocumentaçãoDocumentação(Procedimento Operacional Padrão)(Procedimento Operacional Padrão)
• Devem estar disponíveis POPs e registros das ações desenvolvidas em relação aos seguintes aspectos: • Qualificação de equipamentos; • Aparelhos analíticos e calibração; • Manutenção, limpeza e sanitização; • Dados pessoais, inclusive qualificação, treinamento,
vestuário e higiene; • Monitoramento ambiental; • Controle de pragas; • Reclamações; • Recolhimento; • Devoluções.

DocumentaçãoDocumentação(Procedimento Operacional Padrão)(Procedimento Operacional Padrão)
• Os livros de registros diários devem ser mantidos junto aos principais equipamentos, e devem registrar, de sua utilização, validações, calibrações, manutenção, limpeza ou operações de reparo, inclusive as datas e a identificação da pessoa que as tenha realizado.

Boas Práticas de Boas Práticas de ProduçãoProdução

ProduçãoProdução
• As operações de produção devem seguir Procedimentos Operacionais Padrão (POP) claramente definidos e aprovados, em conformidade com o Relatório Técnico aprovado quando da concessão do registro sanitário, com o objetivo de que sejam obtidos produtos que estejam dentro dos padrões de qualidade exigidos.

Produção (cont.)Produção (cont.)
• Manipulações de materiais e de produtos, tais como o recebimento, quarentena, amostragem, armazenamento, suprimento, processamento, rotulagem, embalagem e distribuição devem ser realizadas de acordo com procedimentos e instruções estabelecidos e registrados.
• Caso ocorram desvios, devem os mesmos serem aprovados por escrito por pessoa designada para tal, com a participação do departamento de controle da qualidade, quando necessário.

Produção (cont.)Produção (cont.)
• Operações realizadas em produtos distintos não devem ser executadas de forma simultânea ou consecutiva na mesma sala, a não ser, que se demonstre que não existe risco de mistura ou de contaminação cruzada.

Produção (cont.)Produção (cont.)
• Durante todo o tempo de processamento, os materiais, recipientes com produtos a granel, os equipamentos maiores e as salas utilizadas devem estar identificadas com etiquetas ou de qualquer outra forma identificação, de modo que seja identificado o produto ou o material que está sendo processado, sua concentração (quando aplicável), e o número do lote.

Produção (cont.)Produção (cont.)
• O acesso às instalações de produção deve ser restrito ao pessoal autorizado.
• Os produtos não farmacêuticos, não devem ser produzidos em áreas ou com equipamentos destinados à produção de medicamentos.

Produção Produção (Prevenção de contaminação cruzada e (Prevenção de contaminação cruzada e contaminação bacteriana na produção)contaminação bacteriana na produção)
• Áreas segregadas• Antecâmaras com diferenciais de pressão de ar e exaustão • Tratamento do ar de entrada• Roupas protetoras nas áreas onde estejam sendo
processados produtos que apresentem risco especial de contaminação cruzada
• Procedimentos de limpeza e de descontaminação de eficácia comprovada
• “sistema de produção fechado”; • Ensaio de resíduos; • Rótulos indicando o estado de limpeza nos equipamentos.

Produção Produção (Operações de processamento: produtos (Operações de processamento: produtos
intermediários e granel)intermediários e granel)
• Mesmos procedimentos e operações de produto acabado.

Produção Produção (Operações de embalagem)(Operações de embalagem)
• Produtos diferentes não devem ser embalados perto uns dos outros, a menos que haja separação física ou sejam aplicados métodos de vigilância eletrônica.
• A liberação da linha de embalagem deve ser feita mediante uma inspeção apropriada e deve ser registrada.
• O nome e número de lote do produto em processo deve ser exibido em cada etapa de embalagem ou na linha de embalagem.

Produção Produção (Operações de embalagem)(Operações de embalagem)
• A conferência de todos os rótulos dentro da linha de O controle em processo de produto durante a embalagem deve incluir, pelo menos, a verificação dos seguintes aspectos: • Aspecto geral• Se as embalagens estão completas; • Utilização de produtos e materiais de
embalagem corretos; • Se as impressões realizadas estão corretas; • Funcionamento correto dos monitores de
processo da linha de embalagem.

Produção Produção (Operações de embalagem)(Operações de embalagem)
• Amostras retiradas do processo de embalagem não devem retornar à linha de embalagem.
• Materiais de embalagem codificados com o número de lote que não forem utilizados devem ser destruídos, devendo o processo de destruição ser registrado. Para que os materiais impressos não codificados sejam devolvidos ao estoque, devem ser seguidos procedimentos estabelecidos.

Boas Práticas de Boas Práticas de Controle de QualidadeControle de Qualidade

Controle de QualidadeControle de Qualidade
• O controle da qualidade é responsável pelas atividades referentes à amostragem, às especificações e aos ensaios, bem como à organização, à documentação e aos procedimentos de liberação que garantam que os ensaios necessários. A independência do controle de qualidade da produção é considerada fundamental.

Controle de QualidadeControle de Qualidade(Controle das matérias primas, (Controle das matérias primas,
produtos intermediários, granel e produtos intermediários, granel e acabados)acabados)• Instruções estabelecidas pelos procedimentos
escritos e aprovados para cada material ou produto• As amostras devem ser representativas dos lotes • Cada recipiente contendo amostra deve conter as
seguintes informações: • nome do material amostrado; • número do lote; • número do recipiente amostrado; • assinatura da pessoa que tenha coletado a
amostra; e • data em que a amostra tenha sido colhida.

Controle de QualidadeControle de Qualidade(Ensaios necessários - matérias (Ensaios necessários - matérias
primas e materiais de embalagem)primas e materiais de embalagem)• Testes quanto à conformidade com as especificações de
identificação, potência, pureza e outros parâmetros de qualidade.
• Ensaios de identificação nas amostras retiradas de cada recipiente de matéria-prima
• Cada lote de material impresso a ser utilizado no processo de embalagem deve ser examinado após o recebimento.
• Aceita-se um certificado de análise emitido pelo fornecedor, desde que a sua confiabilidade seja estabelecida através da validação periódica dos resultados apresentados e através de auditorias à suas instalações

Controle de QualidadeControle de Qualidade(Controle em processo)(Controle em processo)
• Devem ser mantidos registros de controle em processo, os quais devem fazer parte do registro dos lotes.

Controle de QualidadeControle de Qualidade(Produto acabado)(Produto acabado)
• Antes de serem liberados os lotes deve-se, determinar mediante ensaios laboratoriais em conformidade com às especificações estabelecidas
• Produtos que não atenderem às especificações estabelecidas, ou a qualquer outro critério de qualidade importante, devem ser reprovados. Se viável, podem ser reprocessados; porém, os produtos reprocessados devem atender a todas as especificações e critérios de qualidade antes de serem aprovados e liberados.

Controle de QualidadeControle de Qualidade(Revisão dos registros de produção)(Revisão dos registros de produção)
• Os registros de produção e de controle devem ser revisados, se determinado lote não atende às especificações
• As amostras retidas de cada lote do produto acabado devem ser mantidas por, pelo menos, um ano após a data de vencimento
• As amostras de princípios ativos devem ser retidas por, pelo menos, um ano após o vencimento dos prazos de validade dos produtos finais aos quais tenham dado origem
• As amostras de materiais e produtos retidos devem ter tamanho suficiente para possibilitar que sejam realizadas, pelo menos, duas reanálises completas

Controle de QualidadeControle de Qualidade(Estudo de estabilidade)(Estudo de estabilidade)
• Avaliar a qualidade e a estabilidade dos medicamentos acabados e, quando necessário, das matérias-primas e dos produtos intermediários.
• O controle da qualidade deve fixar as datas de vencimento e as especificações quanto ao prazo de validade, tendo como base os ensaios de estabilidade realizados de acordo com as condições de armazenamento.

Controle de QualidadeControle de Qualidade(Estudo de estabilidade)(Estudo de estabilidade)
• Programa escrito e permanente de determinação da estabilidade deve ser desenvolvido e implementado incluindo os seguintes elementos: • Descrição completa do medicamento envolvido no estudo• Parâmetros dos métodos e ensaios, que devem descrever os
procedimentos dos ensaios de potência e de pureza e as características físicas, bem como as evidências documentadas que os ensaios realizados são indicadores da estabilidade do produto
• Previsão quanto à inclusão de um número suficiente de lotes • Cronograma de ensaio para cada medicamento• Instruções sobre condições especiais de armazenamento• Instruções quanto à retenção adequada de amostras• Resumo de todos os dados gerados, incluindo a avaliação e as
conclusões do estudo

Controle de QualidadeControle de Qualidade(Estudo de estabilidade)(Estudo de estabilidade)
• A estabilidade de um produto deve ser determinada antes da comercialização e
deve repetir-se após quaisquer mudanças significativas nos processos de produção, equipamentos, materiais
de embalagem, etc.

Controle de Controle de AlteraçõesAlterações
“Change Control”“Change Control”

Toda e qualquer alteração deve ser
previamente estudada e
autorizada pelo responsável, devendo ser
contemplada com procedimento
específico

DistribuiçãoDistribuição

DISTRIBUIDORADISTRIBUIDORA
Nos registros dedistribuição dos
produtos deverãoconstar:
DistribuiçãoDistribuição

DISTRIBUIDORADISTRIBUIDORA
Destinatário, data de envio, quantidade,
numero do lote, nomedo produto e descrição
do produto.
DistribuiçãoDistribuição

Desta forma, poderemosmanter os arquivos de dados para investigaçõesfuturas ou se necessáriorastrear o produto.
DISTRIBUIDORADISTRIBUIDORA
DistribuiçãoDistribuição

Os registros deverãoser arquivados por um
mínimo de um ano apósa data de validade do
produto
DISTRIBUIDORADISTRIBUIDORA
DistribuiçãoDistribuição

Medicamentos Medicamentos EstéreisEstéreis


Vamos resumir...Vamos resumir...

10 Princípios 10 Princípios Básicos do Básicos do
cGMP cGMP (“Current Good Manufacturing (“Current Good Manufacturing
Practices”)Practices”)

Princípio #1Princípio #1• Escrever e
elaborar documentosdocumentos, de forma detalhada (“passo a passo”), servindo como guias para um desempenho consistente e controlado.

Princípio #2Princípio #2
•Seguir cuidadosamente os
procedimentos procedimentos escritosescritos

Por que é Por que é tão tão importante importante seguir seguir procedimentprocedimentos escritos os escritos ??

• Manter os padrões de desempenho
• Instruções• Referência• Controle• Revisão• Documentação

Nosso dia a dia é repleto Nosso dia a dia é repleto de procedimentos... de procedimentos...
somos seres com somos seres com “hábitos”...“hábitos”...
A diferença é que não A diferença é que não são “ESCRITOS” !!!!!são “ESCRITOS” !!!!!

Os Os procedimentos procedimentos escritos são escritos são nossos mapas nossos mapas para para atingirmos a atingirmos a
QualidadeQualidade

Princípio #3Princípio #3• Ter sempre todos os
documentos documentos de trabalho disponíveis e cuidadosamente cuidadosamente preenchidospreenchidos, visando estar de acordo com as normas e proporcionando rastreabilidaderastreabilidade quando necessária.

Princípio #4Princípio #4
• Provar que os sistemas fazem
o que foram designados para fazer, através de um trabalho de
validaçãovalidação.

Princípio #5Princípio #5• Integrar a
produtividadeprodutividade, a qualidadequalidade do produto e a segurançasegurança do operador quando planejar áreas áreas produtivasprodutivas ou utilização de equipamentosequipamentos.

Princípio #6Princípio #6
• Manter de forma adequada a
manutençãomanutenção de instalações e equipamentos produtivos.

Princípio #7Princípio #7
• Possuir definições claras de desenvolvimento pessoal e descrições de descrições de cargoscargos na empresa.

Princípio #8Princípio #8
Promover a proteção dos produtos contra contaminações, fazendo da
limpeza e limpeza e higienehigiene um hábito diário.

Princípio #9Princípio #9• Qualidade traduzida
em produtos - utilização de
controlescontroles sistemáticos de seus ítens e processos de fabricação, embalagem, rotulagem, testes analíticos, distribuição e mercado.

ORDEM DE EMBALAGEMORDEM DE EMBALAGEM
MATERIAL CODIGO QUANT. TEÓRICA UN QTDE REAL
CARTUCHOS
BULAS
RÓTULOS
MATERIAL CODIGO QUANT. REAL
CARTUCHOS
BULAS
RÓTULOS
100,20100,30100,30
ORDEM DE PRODUÇÃO Princípio Princípio #10#10• Conduzir
auditorias e auditorias e inspeçõesinspeções periódicas para aderência e desempenho segundo normas estabelecidas.

Exercício IVExercício IV
Seus conceitos mudaram?Vamos conferir...

Quais as Quais as consequências da consequências da
inexistência de uma inexistência de uma Estratégia de Estratégia de QualidadeQualidade ?? ??

... Má Qualidade !... Má Qualidade !

Perdas mensuráveis...Perdas mensuráveis...• Erros• Retrabalho• Verificação• Paradas,
atrasos no processo
• Reclamações dos clientes
25% do faturament
o em média

E as perdas não E as perdas não mensuráveis?mensuráveis?
• Quanto custa um funcionário desmotivado?
• Quanto a empresa perde por não aproveitar a criatividade de seus funcionários?
• Quais as consequências de um ambiente de trabalho sujo e desorganizado?

E as perdas não E as perdas não mensuráveis?mensuráveis?
• Quanto custa um cliente insatisfeito?
• Quanto custa reconquistar um cliente insatisfeito?
• Quantos negócios são perdidos devido a algum problema de qualidade anterior?

De onde vêm as perdas?De onde vêm as perdas?
• Equipamento obsoleto• Instruções de trabalho inexistentes ou
inadequadas• Falta de treinamento em todos os níveis• Tecnologia ultrapassada• Métodos de gerenciamento
ultrapassados

• Problemas de comunicação com/entre funcionários e gerentes
• Falta de meios para coletar e implementar sugestões dos funcionários
• Falta de definição clara das responsabilidades
• Centralização excessiva das decisões• Não utilização de métodos adequados
para melhoria das atividades

Mensagens finais...

Utilize sempre linguagem simples e terminologias
claras....

Não “complique” seus procedimentos, escreva “exatamente” como é
feito...

Justifique suas ações e controle os gastos...

Esclareça todas as suas dúvidas e “mitos” sobre
cGMP/BPF - isto pode custar uma fortuna à sua
empresa ...

Lembre-se de que toda alteração de controle deve ser gerenciada e documentada ...

Estimule debates ... Qual é o custo efetivo da
Qualidade ?

Acima de tudo...Mantenha o senso crítico e
de proporção !!!

Mas o que é mesmo o Mas o que é mesmo o cGMP?cGMP?

Exercício VExercício V

....................................................
.................... ....................
....................... .......................

Perguntas ?Perguntas ?

Referências para consultas Referências para consultas futuras:futuras:
• FDA - www.fda.gov• Anvisa - www.anvisa.gov.br• www.gmp1st.com• CD Rom - HSV Consultores
Associados: – Cópia da apresentação – Norma BPF - Brasil – GMP Guide


















