
dissertação Rev Final
201
1 CAPÍTULO 1 1.1 Introdução Como indica o tema da presente dissertação, os processos corrosivos e os associados à fragilização do aço, os quais, com o tempo, se estabelecem nos componentes dos equipamentos de processos tipicamente encontrados em refinarias de petróleo, constitui parte central da mesma. A fragilização, que se expressa por uma degradação das propriedades originais do material, e que se deve a uma exposição contínua a elevadas ou a baixas temperaturas e/ou a meios promotores do fenômeno, pode ou não vir acompanhado por solicitações mecânicas significativas. De modo similar, o processo corrosivo, seja ele desenvolvido em meio aquoso ou à temperaturas superiores à do ponto de orvalho do ambiente em questão, decorre da interação do metal com o meio no qual encontra-se imerso. Desta forma, o conhecimento ainda que apenas superficial do refino e de seus vários processos, e dos vários ambientes ou meios a eles associados, juntamente com a metalurgia mais costumeiramente empregada naquelas posições, são absolutamente necessários se se deseja antecipar o mecanismo de corrosão operante nos vários trechos dessas plantas. Evidentemente, tal conhecimento reveste-se da mais alta importância relativamente à confiabilidade das instalações. Além da antecipação de problemas, permite, numa fase inicial de projeto, selecionar ligas metálicas que apresentem uma melhor relação custo/benefício para uma dada aplicação, tipicamente um aço e freqüentemente o aço selecionado é o aço carbono ou carbono-manganês, nas suas mais variadas especificações. Numa fase subseqüente, estando a planta operando, é importante monitorar e tomar ações no sentido de mantê-lo controlado ao se constatar, por qualquer razão, o recrudescimento da corrosividade. É, da mais alta importância, ainda, selecionar pontos para uma investigação (inspeção) mais minuciosa e especificar a técnica de ensaio não destrutivo mais apropriada, nas paradas de manutenção previamente programadas. Deverá servir também para proceder análises de falhas ocorridas em serviço, infelizmente não raras, podendo estar associadas a um significativo impacto financeiro, à saúde do trabalhador e ao meio ambiente. Como resultado, o conjunto de informações reunidas nessa dissertação deve permitir tecer
Transcript of dissertação Rev Final

1 CAPÍTULO 1
1.1 Introdução
Como indica o tema da presente dissertação, os processos corrosivos e os associados à
fragilização do aço, os quais, com o tempo, se estabelecem nos componentes dos
equipamentos de processos tipicamente encontrados em refinarias de petróleo, constitui
parte central da mesma.
A fragilização, que se expressa por uma degradação das propriedades originais do
material, e que se deve a uma exposição contínua a elevadas ou a baixas temperaturas
e/ou a meios promotores do fenômeno, pode ou não vir acompanhado por solicitações
mecânicas significativas. De modo similar, o processo corrosivo, seja ele desenvolvido
em meio aquoso ou à temperaturas superiores à do ponto de orvalho do ambiente em
questão, decorre da interação do metal com o meio no qual encontra-se imerso. Desta
forma, o conhecimento ainda que apenas superficial do refino e de seus vários
processos, e dos vários ambientes ou meios a eles associados, juntamente com a
metalurgia mais costumeiramente empregada naquelas posições, são absolutamente
necessários se se deseja antecipar o mecanismo de corrosão operante nos vários trechos
dessas plantas.
Evidentemente, tal conhecimento reveste-se da mais alta importância relativamente à
confiabilidade das instalações. Além da antecipação de problemas, permite, numa fase
inicial de projeto, selecionar ligas metálicas que apresentem uma melhor relação
custo/benefício para uma dada aplicação, tipicamente um aço e freqüentemente o aço
selecionado é o aço carbono ou carbono-manganês, nas suas mais variadas
especificações. Numa fase subseqüente, estando a planta operando, é importante
monitorar e tomar ações no sentido de mantê-lo controlado ao se constatar, por qualquer
razão, o recrudescimento da corrosividade. É, da mais alta importância, ainda,
selecionar pontos para uma investigação (inspeção) mais minuciosa e especificar a
técnica de ensaio não destrutivo mais apropriada, nas paradas de manutenção
previamente programadas. Deverá servir também para proceder análises de falhas
ocorridas em serviço, infelizmente não raras, podendo estar associadas a um
significativo impacto financeiro, à saúde do trabalhador e ao meio ambiente. Como
resultado, o conjunto de informações reunidas nessa dissertação deve permitir tecer

2 recomendações que possam prevenir repetições das falhas e, em paralelo, sendo o
caso, retro-alimentar o projeto.
Assim sendo, nessa dissertação, o objetivo é dar ênfase tanto à caracterização do
ambiente existente em alguns dos processos mais comumente encontrados em
refinarias, como à metalurgia tipicamente empregada nestas plantas e em posições
específicas, e daí, ao processo corrosivo/fragilização propriamente dito. Por
conseqüência, a adoção de ações mitigadoras, particularmente aquelas associadas com a
atenuação da agressividade do meio, bem como recomendações da metalurgia, deverão
decorrer dessas considerações.
A maioria dos casos de corrosão ou de deterioração considerados neste trabalho, são
oriundos de ocorrências sofridas pelos mais variados tipos de equipamentos, existentes
nas unidades de processamento e de tratamento de derivados da Refinaria Gabriel
Passos (REGAP). Alguns outros poucos casos oriundos da literatura especializada, cujo
registro inexiste nos arquivos da refinaria, também constituem este trabalho.
Presentemente, a REGAP processa cerca de 150.000 barris de petróleo por dia (∼ 1.000
m3/h), todo ele oriundo da bacia de Campos. Destila, craqueia, produz coque e
derivados leves a partir de resíduos pesados provenientes da destilação atmosférica, da
destilação a vácuo e do coqueamento retardado. A REGAP também desnitrifica e
dessulfuriza algumas de suas principais frações observando questões ambientais, o que
lhe permite abastecer grande parte dos centros consumidores do estado de Minas Gerais
(∼ 70%), e também o Distrito Federal, Goiânia, Anápolis e Vitória, encontrando-se em
operação a partir do início de 1968.

3 CAPÍTULO 2
2.1 O petróleo
Oriundo da decomposição de animais e vegetais aquáticos soterrados por sucessivas
camadas de material sedimentar, sob condições em que a ação bacteriana prevaleceu à
oxidação, isto é, em um ambiente pressurizado, aquecido e virtualmente isento de
oxigênio, o petróleo é assim essencialmente constituído por material de origem
orgânica. [1, 2]
Entre várias outras características, suas moléculas constituintes, os hidrocarbonetos, são
estruturados por covalência através de ligações dos tipos simples C−C, dupla C=C
(apolares) e C−H (fracamente polarizado). Tais moléculas são mantidas unidas entre si
por forças intermoleculares, F, de Van der Walls, F = –dU/dr ∝ r–7 → F ∝ r–6 , onde U
simboliza a energia de ligação entre moléculas vizinhas e r a distância entre essas
mesmas moléculas [1]. Portanto, a intensidade da interação entre moléculas próximas
reduz-se de forma muito rápida com a distância. Entretanto, F tende a aumentar com o
volume das mesmas, devido ao aumento da quantidade de elétrons capazes de interagir,
bem como com a redução da influência da carga do núcleo sobre esses mesmos elétrons.
Possuem, grosso modo, uma viscosidade absoluta variando entre cerca de 3×10−4 a
5×10−1Pa.s, densidade d entre ~ 0,8 (°API = 141,5
d −131,5 ≅ 45) e ~ 0,9 (°API ≅ 26) à
temperatura ambiente. Formam solventes fortemente apolares e, como tais, são
insolúveis nos solventes polares como são a água e o álcool. Evidentemente, um
petróleo mais leve, ou de maior grau API, apresenta valor comercial superior por ser
mais facilmente processado e rico nos componentes mais valorizados pelo mercado de
derivados (GLP, gasolina e diesel). [3]
Mais especificamente, o petróleo é composto por uma mistura liqüefeita de
hidrocarbonetos possuidores de diferentes massas molares, assim classificados: alcanos
(são os hidrocarbonetos ″parafínicos″; cadeia acíclica ramificada ou não, saturado),
ciclo-alcanos (são os hidrocarbonetos ″naftênicos″ ou ciclo-parafínicos; cadeia cíclica
ramificada ou não, saturado), e os aromáticos (são os hidrocarbonetos estruturados por
anéis benzênicos, não-saturados mas possuidores de elevada estabilidade). Pela
importância, deve-se citar ainda os alcenos (são hidrocarbonetos ″olefínicos″; cadeias
4 acíclicas e insaturadas, portanto, fortemente reativos), os quais formam um grupo de
hidrocarbonetos unicamente encontrados nos produtos que resultam da operação de
craqueamento catalítico [1, 3].
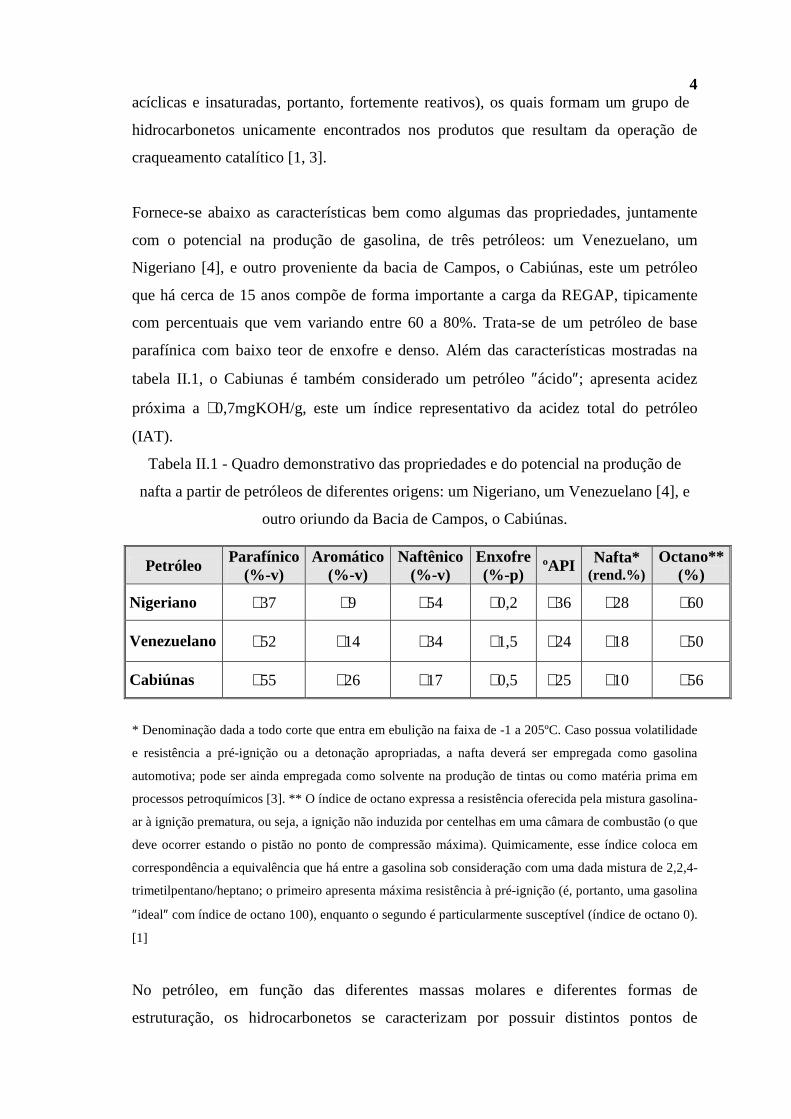
Fornece-se abaixo as características bem como algumas das propriedades, juntamente
com o potencial na produção de gasolina, de três petróleos: um Venezuelano, um
Nigeriano [4], e outro proveniente da bacia de Campos, o Cabiúnas, este um petróleo
que há cerca de 15 anos compõe de forma importante a carga da REGAP, tipicamente
com percentuais que vem variando entre 60 a 80%. Trata-se de um petróleo de base
parafínica com baixo teor de enxofre e denso. Além das características mostradas na
tabela II.1, o Cabiunas é também considerado um petróleo ″ácido″; apresenta acidez
próxima a ∼0,7mgKOH/g, este um índice representativo da acidez total do petróleo
(IAT).
Tabela II.1 - Quadro demonstrativo das propriedades e do potencial na produção de
nafta a partir de petróleos de diferentes origens: um Nigeriano, um Venezuelano [4], e
outro oriundo da Bacia de Campos, o Cabiúnas.
Petróleo Parafínico (%-v)
Aromático (%-v)
Naftênico (%-v)
Enxofre (%-p)
ºAPI Nafta* (rend.%)
Octano** (%)
Nigeriano ∼37 ∼9 ∼54 ∼0,2 ∼36 ∼28 ∼60
Venezuelano ∼52 ∼14 ∼34 ∼1,5 ∼24 ∼18 ∼50
Cabiúnas ∼55 ∼26 ∼17 ∼0,5 ∼25 ∼10 ∼56
* Denominação dada a todo corte que entra em ebulição na faixa de -1 a 205ºC. Caso possua volatilidade
e resistência a pré-ignição ou a detonação apropriadas, a nafta deverá ser empregada como gasolina
automotiva; pode ser ainda empregada como solvente na produção de tintas ou como matéria prima em
processos petroquímicos [3]. ** O índice de octano expressa a resistência oferecida pela mistura gasolina-
ar à ignição prematura, ou seja, a ignição não induzida por centelhas em uma câmara de combustão (o que
deve ocorrer estando o pistão no ponto de compressão máxima). Quimicamente, esse índice coloca em
correspondência a equivalência que há entre a gasolina sob consideração com uma dada mistura de 2,2,4-
trimetilpentano/heptano; o primeiro apresenta máxima resistência à pré-ignição (é, portanto, uma gasolina
″ideal″ com índice de octano 100), enquanto o segundo é particularmente susceptível (índice de octano 0).
[1]
No petróleo, em função das diferentes massas molares e diferentes formas de
estruturação, os hidrocarbonetos se caracterizam por possuir distintos pontos de
5 ebulição, os quais aumentam com o aumento do número de átomos de carbono na
molécula (ou com a massa molar), em uma mesma série homóloga. Não obstante
apresentar-se líquido sob condições ordinárias de pressão e de temperatura, o petróleo
mantém dissolvidos hidrocarbonetos com muito baixos pontos de ebulição (voláteis),
gasosos nessas condições, bem como hidrocarbonetos com elevados pontos de ebulição;
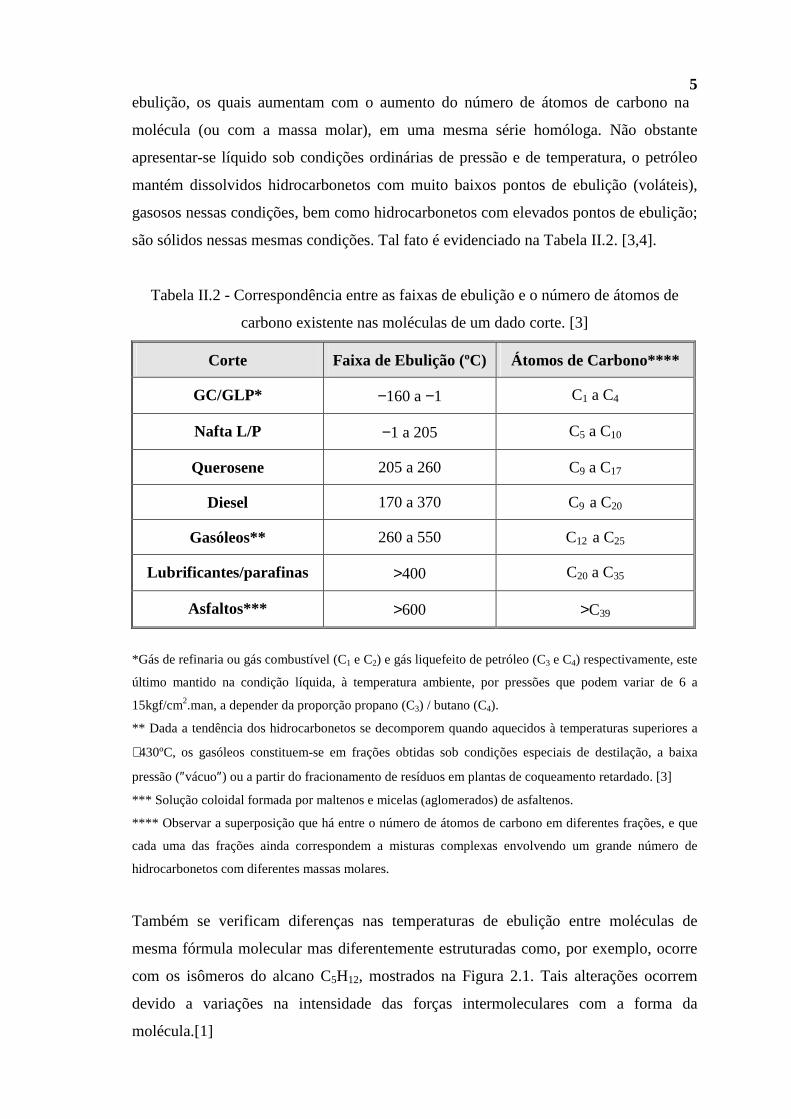
são sólidos nessas mesmas condições. Tal fato é evidenciado na Tabela II.2. [3,4].
Tabela II.2 - Correspondência entre as faixas de ebulição e o número de átomos de
carbono existente nas moléculas de um dado corte. [3]
Corte Faixa de Ebulição (ºC) Átomos de Carbono****
GC/GLP* −160 a −1 C1 a C4
Nafta L/P −1 a 205 C5 a C10
Querosene 205 a 260 C9 a C17
Diesel 170 a 370 C9 a C20
Gasóleos** 260 a 550 C12 a C25
Lubrificantes/parafinas >400 C20 a C35
Asfaltos*** >600 >C39
*Gás de refinaria ou gás combustível (C1 e C2) e gás liquefeito de petróleo (C3 e C4) respectivamente, este
último mantido na condição líquida, à temperatura ambiente, por pressões que podem variar de 6 a
15kgf/cm2.man, a depender da proporção propano (C3) / butano (C4).
** Dada a tendência dos hidrocarbonetos se decomporem quando aquecidos à temperaturas superiores a
∼430ºC, os gasóleos constituem-se em frações obtidas sob condições especiais de destilação, a baixa
pressão (″vácuo″) ou a partir do fracionamento de resíduos em plantas de coqueamento retardado. [3]
*** Solução coloidal formada por maltenos e micelas (aglomerados) de asfaltenos.
**** Observar a superposição que há entre o número de átomos de carbono em diferentes frações, e que
cada uma das frações ainda correspondem a misturas complexas envolvendo um grande número de
hidrocarbonetos com diferentes massas molares.
Também se verificam diferenças nas temperaturas de ebulição entre moléculas de
mesma fórmula molecular mas diferentemente estruturadas como, por exemplo, ocorre
com os isômeros do alcano C5H12, mostrados na Figura 2.1. Tais alterações ocorrem
devido a variações na intensidade das forças intermoleculares com a forma da
molécula.[1]

6
Figura 2.1: Isômeros estruturais do pentano com seus diferentes pontos de ebulição. [1]
2.1.1 Processamento do petróleo. Generalidades
Com unicamente três exceções, sendo apenas uma unidade (Lubnor-CE) dedicada
integralmente à produção de lubrificantes, a maior parte das refinarias que compõem o
parque de refino da Petrobras no Brasil (oito de um total de dez refinarias), objetiva,
predominantemente, o processamento do petróleo visando a produção de combustíveis
(GLP, gasolina, querosene para aviação ou QAV e diesel). Entre essas oito refinarias
encontra-se a própria REGAP. Assim, seu ″esquema de refino″, ou seja, as
características e a interligação entre as várias unidades de processo existentes na
refinaria levam em conta essa ″filosofia″ produtiva. Contudo, prevê-se em futuro
próximo, a incorporação de mais uma unidade, uma unidade ″separadora de propeno″,
para atender as necessidades do também futuro, ″complexo acrílico″, a ser instalado nas
vizinhanças da REGAP. Tal complexo visará a produção do ácido acrílico e daí de
acrilatos, estes empregados na obtenção de tintas acrílicas e materiais absorventes, por
exemplo.
Diferenças nas propriedades físicas viabilizam a separação dos hidrocarbonetos sem que
seja necessário proceder alterações na natureza química da molécula. Em uma refinaria,
tais metodologias compõem os ″processos de separação″, os quais, por sua vez,
objetivam desdobrar o petróleo em frações, ou extrair de uma dada fração um grupo
específico de compostos [5].
Relativamente aos processos de separação, destaca-se a ″destilação fracionada″,
metodologia que faz uso de temperaturas e pressões tais que a separação se dá com base
nas diferenças existentes entre os pontos de ebulição dos componentes individuais da
mistura líquida. Então, além da temperatura, a pressão se constitui numa importante

7 variável do processo. Isso ocorre pelo fato de que o ponto de ebulição de um dado
hidrocarboneto se altera em função da pressão sobre ele exercida pelo ambiente.
Exemplificando, o ponto de ebulição do n-pentano é de 36°C porque nessa temperatura
sua pressão de vapor se equilibra com a pressão atmosférica, ∼101,3kPa. Então, a
redução dessa pressão implicaria na redução do ponto de ebulição do n-pentano e vice-
versa. Assim, recorre-se ao artifício da redução da pressão quando se deseja fracionar
″resíduos″ cujos componentes possuam pontos de ebulição superiores ao de sua
decomposição térmica, o que se dá em torno de 430ºC. Distinguem-se, assim, a
″destilação atmosférica″, na qual o fracionamento do petróleo é obtido numa coluna ou
"torre atmosférica" com temperaturas variando entre ~120 (topo) a ~380°C (fundo) à
pressão atmosférica. Nestas condições, separam-se GLP pelo topo da torre, nafta,
querosene e diesel em cortes laterais, e pelo fundo, obtém-se um ″resíduo atmosférico″
(RAT), que segue como carga da ″destilação a vácuo" [3, 5].
De uma forma geral, esse resíduo e os demais que resultam do fracionamento do
petróleo, são constituídos por uma fase dispersa sob a forma de micelas composta por
″asfaltenos″ de elevada massa molar contendo quantidades significativas de metais,
enxofre e nitrogênio, e de uma fase orgânica dispersora com menor massa, os
″maltenos″, esses uma mistura complexa constituída por hidrocarbonetos saturados e
aromáticos, também contendo metais, Figura 2.2. [6, 7]
Figura 2.2: Estrutura de um asfalteno. A parte aromática encontra-se interligada através
de cadeias alifáticas, pontes de enxofre e por moléculas de porfirina
contendo íons de níquel e vanádio em seu núcleo. [6]

8 Entre os efeitos deletérios causados pelos asfaltenos, pode-se citar sua grande
propensão à formação de resíduos carbonáceos sólidos (coque) via degradação térmica,
que poderia levar, por exemplo, à formação de pontos quentes nas "serpentinas" dos
fornos de processo. Em correspondência, a fração do coque gerado por decomposição
dos asfaltenos existentes no RAT de um petróleo brasileiro 19°API, atingiu o valor de
49% ao passo que o mesmo resíduo isento de asfaltenos contribuiu com apenas 4% [7].
Além da formação de pontos quentes, os asfaltenos podem acarretar a desativação de
catalisadores das plantas de craqueamento. Os metais contidos nos asfaltenos tendem a
se acumular e obstruir os poros dos catalisadores; o coque deles gerado pode recobrí-los
totalmente. Além disso, os asfaltenos podem gerar compostos agressivos ao aço tais
como são o H2S, o V2O5, e o HCN.
Em uma destilação a vácuo, o fracionamento é conduzido numa coluna ou "torre
vácuo" à pressões que variam entre cerca de 100 a 10 mmHg (fundo e topo da torre,
respectivamente), o que acarreta uma redução da ordem de 150ºC nos pontos de
ebulição do hidrocarboneto. Nesta torre, na qual não existe retirada de topo, havendo
apenas a extração de vapor d’água em mistura com hidrocarbonetos leves e
contaminantes inorgânicos arrastados devido ao efeito de sucção ocasionado pelo
sistema de vácuo, obtém-se, via retiradas laterais, os gasóleos leve (∼diesel) e pesado
(GOL e GOP, respectivamente). Esse último que seria empregado como óleo
combustível, é carga da unidade de "craqueamento catalítico fluido", e pelo fundo,
obtém-se um novo resíduo, o ″resíduo de vácuo″ (RV), que segue como carga da planta
de "coqueamento retardado". Assim, na medida que processam resíduos, essas duas
últimas unidades desempenham um papel fundamental na rentabilidade de uma
refinaria.
Além da temperatura e pressão, uma ″separação″ pode envolver também o emprego de
solventes ou o uso de materiais adsorventes, chamadas ″peneiras moleculares″. Tais
métodos visam a extração de compostos específicos de uma dada carga de
hidrocarbonetos. Servem de exemplo, a extração de compostos aromáticos para o
enquadramento da viscosidade de óleos lubrificantes ou de n-parafinas para a redução
do ponto de congelamento do querosene. É o caso das plantas de ″desaromatização a
furfural″ e as de ″adsorção de n-parafinas″, respectivamente [5]. Contudo, neste
trabalho, em razão da inexistência de plantas desses tipos na REGAP e também devido

9 a grande importância que as destilações assumem nos esquemas de refino, o enfoque
se limitará aos processos corrosivos tipicamente encontrados nas destilações
atmosférica e vácuo.
Em adição aos métodos de separação, o processamento do hidrocarboneto pode
envolver ″conversões″. Diferentemente do caso anterior, uma conversão envolve
alterações irreversíveis da natureza química da molécula. Objetiva, sempre, a obtenção
de produtos com maior valor agregado a partir de outros com menor valor (por
exemplo, ao enviar o GOP para o craqueamento, tal como mencionado acima). Isto
pode ser conseguido através de reações que acarretam a ruptura, o reagrupamento ou a
reestruturação molecular, que pode ou não ser assistido por catalisadores [5].
Pode-se citar como exemplo de uma conversão catalítica de grande importância, o
craqueamento ou a ruptura provocada em moléculas de hidrocarbonetos pesados em
leito de catalisador, trata-se de um material granulado com elevada área superficial, a
base de SiO2 e Al2O3, que é mantido “fluidizado”, ou em suspensão, numa massa de
hidrocarbonetos vaporizada, à temperaturas relativamente elevadas (~650°C). Tal
processo se passa ao longo de uma tubulação vertical (″riser″), estando a mistura
vaporizada em escoamento ascendente, e se completa num reator posicionado em sua
extremidade superior. No reator, por se promover uma abrupta redução de velocidades,
tem início também a separação das partículas do catalisador do hidrocarboneto
craqueado, separação que é complementada em um conjunto de ″ciclones″ por meio de
efeito centrífugo. Essa operação é levada a efeito em ″unidades de craqueamento
catalítico fluido″ (UFCC), e produz, como efluentes gasosos os hidrocarbonetos C1 a C4
a partir de cargas de valor comercial muito baixo (GOP). A corrente, proveniente do
reator, é enviada para uma torre fracionadora, onde se separam os leves pelo topo da
torre (GC, GLP e nafta), do hidrocarboneto mais pesado e apenas parcialmente
craqueado; trata-se do ″óleo de reciclo leve″ (LCO), retirada lateral que segue para o
hidrotratamento como diesel.
Serve ainda como exemplo de um processo de conversão catalítico, a conversão de
alcanos lineares em ramificados (processo denominado ″isomerização″), estes últimos
possuidores de maiores índices de octano. Entretanto, por razões idênticas às apontadas

10 anteriormente, será dado enfoque apenas aos eventos de degradação do aço que se
passa em UFCC’s.
Dada a importância que assume no contexto da presente dissertação, será feita
referência também à fragilização sofrida pelo aço em ″unidades de recuperação de
gases″ (URG), planta que se posiciona imediatamente a jusante do sistema de topo de
uma fracionadora-UFCC. Essa planta tem a função de recolher e separar os
hidrocarbonetos gasosos (GC e GLP) da nafta líquida efluente do topo da torre
fracionadora, e subseqüentemente, o próprio GC do GLP.
Um segundo exemplo de plantas envolvendo mecanismos de conversões, neste caso não
catalítica, também de grande importância econômica em refinarias e que deverá ser
considerado nesta dissertação, diz respeito a planta de produção de "coque de petróleo",
juntamente com diesel, nafta e GLP. O coque é constituído por cadeias poliméricas
sólidas, com elevadas massas molares e elevadas razões C/H, cuja principal demanda
dá-se na produção de alumínio como eletrodo de células eletrolíticas. Toda essa
produção ocorre em ″unidades de coqueamento retardado″ (UCR), o que se faz a partir
do craqueamento térmico (a cerca de 490ºC) do resíduo que se produz no fundo da
fracionadora da unidade, que recebe como carga o RV [5]. O termo ″retardado″ decorre
das ações tomadas para evitar que a formação do coque se dê nos tubos da fornalha, há
injeção de água de caldeira para promover turbulência naquelas posições, retardando
essa formação de forma que ela venha a ocorrer no local apropriado, o ″tambor ou
reator de coque″.
Como exemplos de processos de tratamento de derivados encontram-se aqueles que
visam a redução dos compostos sulfurados e nitrogenados por questões ambientais, mas
também para conferir estabilidade química, eliminação de odores e redução da
corrosividade, selecionou-se uma unidade de tratamento com aminas (DEA) e de
hidrotratamento (HDT). A primeira unidade faz uso de solução aquosa líquida
absorvente de H2S/CO2, sendo apropriada para dessulfurizar hidrocarbonetos gasosos
(GC e GLP). A segunda unidade é empregada na dessulfurização e desnitrificação de
frações médias e pesadas (nafta, querosene e diesel), empregando como insumo gás
hidrogênio, que é produzido na própria refinaria em unidades de geração de hidrogênio

11 (UGH) a partir da reforma a vapor do gás natural ou nafta, planta que compõe um
serviço ″auxiliar″ que também é objeto de considerações neste trabalho.
2.1.2 A ″″″″corrosividade" do petróleo
Dada sua natureza apolar, os hidrocarbonetos não interagem quimicamente com
sistemas de ligas metálicas. Não são, portanto, corrosivos. Essa é uma afirmativa que
vale para ambos: o petróleo e seus derivados. No entanto, tanto um como o outro
contém sempre, como contaminantes, em maior ou menor extensão, além dos
compostos inorgânicos, compostos orgânicos sulfurados, nitrogenados, oxigenados e
organo-metálicos, esses contendo os metais níquel e vanádio principalmente. Uma faixa
de composições (%-p), caracteristicamente encontradas nos petróleos é apresentada na
Tabela II.3.
Tabela II.3 - Composição química típica dos petróleos. [4]
C H N O S Ni-V
83,0-87,0 10,0-14,0 0,1-2,0 0,05-1,5 0,05-6,0 <1000ppm
Compostos sulfurados
A concentração dos compostos sulfurados orgânicos tende a intensificar-se nos
petróleos mais pesados com menor °API, e segregar-se nas frações mais pesadas ou
resíduos [4]. Dentre eles, destacam-se o sulfeto de hidrogênio, H2S, os tióis (ou
mercaptans), − SH, os tiofenois, = SH, os sulfetos, − S −, os dissulfetos −S−S− e ainda o
enxofre elementar, S. A agressividade desses compostos ao aço se reduz na ordem dada,
sendo o H2S o principal responsável pela corrosão por ″sulfetação″, processo que ocorre
numa faixa intermediária de temperaturas (∼260 a ∼400ºC), com um pico de
agressividade a cerca de 370ºC [8]. Em particular, a agressividade dos compostos
sulfurados parece estar, pelo menos em parte, associada ao fato de se decomporem em
H2S com o aumento da temperatura. O enxofre elementar, se presente no óleo, deve
iniciar sua decomposição, S → H2S, a partir de cerca de 150ºC para atingir um máximo
a 220ºC, ao passo que a decomposição dos demais compostos se dá a temperaturas mais
altas [4]. Por outro lado, o H2S solubilizado em água (ou seja, como ácido), proporciona
a fragilização do aço, fenômeno que se inicia pela corrosão do aço. Assim, dada a
importância e a sistemática presença desses compostos, o petróleo é também

12 classificado em função do teor total de enxofre presente. Será um petróleo BTE
(baixo teor de enxofre ou “sweet”) se o enxofre total se limitar a 1%(p), e será um
petróleo ATE (alto teor de enxofre ou “sour”) caso essa limitação não se cumpra.
Compostos oxigenados
Além dos sulfurados, aparece também muito relevantemente no que diz respeito à
corrosão do aço em refinarias na faixa 200-400°C, os compostos orgânicos oxigenados,
responsáveis pela acidez apresentada pelo petróleo. No que diz respeito à corrosão, o
mais importante destes compostos se estrutura sob a forma de uma ″grande família de
ácidos″, onde o grupamento carboxílico se apresenta ligado a aneis ciclo-alcanos,
R(CH2)mCOOH, através de m (≥1) unidades CH2. A massa molar desses compostos
varia entre 200 e 700g/mol, existindo a indicação de que os mais agressivos sejam
aqueles possuidores de menor massa. [8, 9]
Tal como ocorre com os compostos sulfurados e metais nos asfaltenos, esses ácidos
também tendem a concentrar-se nas frações mais pesadas por possuírem pontos de
ebulição mais compatível com o hidrocarboneto presente nessas frações. Um petróleo
será considerado como potencial causador de corrosão naftênica se possuir um IAT
(índice de acidez total determinado por método titulométrico) igual ou superior a
0,5mgKOH/g-amostra, observando-se, desde já, ser da mais alta importância conhecer-
se o modo pelo qual o ácido se distribui nos derivados (IAT’s), e, assim, na planta, a
partir de um dado petróleo ou de misturas de petróleos. [10]
Compostos nitrogenados
Tais compostos têm também sua presença no óleo associada aos asfaltenos (porfirinas).
Assim, tendem a concentrar-se nos resíduos. Além de ocasionar o escurecimento do
diesel e demais frações intermediárias devido a sua reatividade, assumem significativa
importância no contexto da corrosão por darem origem, nas UFCC’s, à amônia e ao
cianeto de hidrogênio, os quais cumprem importante papel na fragilização do aço em
presença do H2S e água. Essa forma de dano ocorre, particularmente, nos equipamentos
que compõem as URG’s. Além da porfirina nos asfaltenos, são exemplos de compostos
nitrogenados encontrados no petróleo: a piridina (C5H5N) e o pirrol (C4H5N). [3]

13 Compostos organo-metálicos
Os metais (vanádio, níquel, ferro e cobre, principalmente) ocasionam a desativação
prematura de catalisadores [3, 4]. Essa é, possivelmente, a mais importante
conseqüência da sua presença no petróleo. Entretanto, por se segregarem nas frações
pesadas e resíduos, tendem a acumular-se no óleo combustível que é empregado nos
fornos de processo. Darão, neste caso, origem a gases de combustão contendo
compostos que, ao se liqüefazerem e interagirem com o aço, proporcionam o “ataque
por cinzas fundidas”, fenômeno que se desenvolve nos acessórios metálicos (suportes,
pendurais etc), existentes nas câmaras de radiação e de convecção desses equipamentos.
Compostos Inorgânicos
Em adição às impurezas orgânicas, o petróleo retém água, sais minerais, areia e argila
arrastados da rocha em que se acumulou, como por exemplo, um arenito, rocha formada
por partículas de sílica (ou areia) aglomeradas por um cimento a base de carbonato. As
impurezas inorgânicas também se originam dos métodos de recuperação que fazem uso
da injeção de salmoura nos poços. [3]. Desses sais, destaca-se o sal hidrolizável MgCl2
o principal gerador de HCl, tornando-se assim, a principal fonte de problemas de
corrosão em baixa temperatura nas destilações nos seus ″sistemas de topo″ [11]. Essa é
uma das razões pelas quais o petróleo deve ser dessalgado em antecedência ao seu
processamento.
2.2 Fluxograma REGAP
A Figura 2.3, apresenta um fluxograma em que se procura sintetizar uma refinaria tal
como se apresenta nos dias de hoje (REGAP), juntamente com alguns dos processos
que são tratados nessa dissertação. Observa-se a sistemática presença de vapor d′água, o
qual viabiliza a maior parte dos processos corrosivos em baixa temperatura. O vapor e,
subseqüentemente, o condensado dele originado, dará origem ao meio aquoso ou
eletrólito promovedor dos processos de corrosão. Nota-se, também, que os principais
compostos corrosivos, alguns deles já citados, bem como os principais cortes e sua
respectiva posição nas plantas, são mostrados. Em alguns casos, tais compostos são
gerados na própria planta por decorrência do processo, em outros são contidos na carga.
São exemplos dos primeiros: o H2SO4 em unidades de recuperação de enxofre (URE), o

14 HCl nas destilações, o NH3 e o H2S nas plantas de hidrotratamento, o HCN no
craqueamento; e como exemplo do segundo, os ácidos carboxílicos ou naftênicos
−COOH e os compostos sulfurados (H2S, −SH), ambos já contidos no petróleo. Não são
indicadas as plantas de ″utilidades″: vapor, ar e água de refrigeração, assim como não
são indicados as de tratamento de águas residuais. (AL = água de lavagem, OC = óleo
combustível, NF = nafta craqueada, GLP = gás liqüefeito de petróleo, GC = gás
combustível, GN = gás natural)
Figura 2.3: Representação esquemática dos principais processos existentes na REGAP,
juntamente com as frações e alguns dos principais compostos agressivos.
2.3 Aços. Características gerais, soldabilidade, corrosão em meio aquoso e exemplos
iniciais de modos de falha encontrados num ambiente de refinarias
Ainda na introdução dessa dissertação, deixou-se antecipado ser o aço carbono, baixo-
médio carbono (%C≤0,35) o material de construção de mais amplo emprego em
refinarias. Pode-se generalizar: são de fato os materiais metálicos de emprego mais
amplo na indústria naval, em plantas químicas ou petroquímicas. Tal aceitação deriva da
excelente relação custo/benefício apresentado por esses materiais. O ″benefício″
decorre, entre várias outras razões, de uma muito boa deformabilidade, de uma razoável
resistência a corrosão às atmosferas úmidas e aos meios aquosos aerados pouco ácidos

15 (pH>5), neutros e alcalinos e de uma adequada soldabilidade. Neste caso, deve-se
considerar o emprego em larga escala da soldagem na fabricação dos mais variados
produtos, incluindo, por exemplo, os ″vasos de pressão″, equipamentos presentes em
grande quantidade numa refinaria. Por soldabilidade entende-se a capacidade do aço em
ter minimamente alteradas as propriedades mecânicas originais e possuir uma baixa
propensão à nucleação de defeitos em sua ″zona termicamente alterada″ (ZTA) por
decorrência de uma soldagem autógena (presumidamente corretamente executada).
A importância da manutenção das propriedades mecânicas nesta posição, propriedades
que podem ser caracterizadas pelos limites de escoamento σy e de resistência σu do aço,
se dá pelo fato de que esses limites servem de referência para o estabelecimento das
tensões admissíveis constantes dos códigos de projeto. Por exemplo, para temperaturas
nas quais a fluência não necessita ser considerada, a tensão admissível que deve
prevenir falhas por ″ruptura dúctil ou por instabilidade plástica″, é segundo o código
ASME, VIII-1 [12], o menor dos valores: σy/1,5 ou σu/3,5. Nesse mesmo contexto,
deve-se considerar ainda a manutenção de uma adequada ductilidade e tenacidade (ao
entalhe ou à fratura). A ductilidade pode ser entendida como sendo a capacidade do
material em redistribuir tensões em pontos onde estiver concentrada, ou ainda, a
capacidade do mesmo em escoar plasticamente antes da ruptura. Por tenacidade à
fratura, entende-se como sendo a capacidade do aço em impor resistência à propagação
sub-crítica de trincas ao plastificar-se ou apresentar-se dúctil na frente de propagação. A
propagação sub-crítica ou estável difere da propagação instável porque essa se processa
a altas taxas independentemente de aumentos no carregamento mecânico. A tenacidade
ao entalhe é indicadora da capacidade do aço em absorver energia proveniente de
impactos (carregamentos exercidos a altas taxas de deformação), particularmente
aqueles sofridos a baixas temperaturas.
Tal como deixou-se indicado acima, os aços carbono em particular, e os aços em geral,
são todos materiais de comportamento elasto-plástico. Um ″comportamento puramente
elástico″ será assegurado desde que o carregamento aplicado σap não leve à geração de
tensões internas que sejam superiores a σy. Nesse sentido, a ″rigidez″ do aço é indicada
pelo valor de seu módulo de elasticidade (E), constante de proporcionalidade que
relaciona a tensão e a deformação elástica εel, ou, σy=Eεel. Um ″comportamento
plástico″, e assim, a plastificação do aço, se seguirá caso não se cumpra σap<σy.

16 Entretanto, neste caso, os aços revelam a existência de um fenômeno (encruamento)
que os tornam mais resistentes à medida que se plastificam; diferentemente do que se
passa no regime elástico, trata-se de um fenômeno de natureza não-linear. Contudo, tal
fato não é (conservativamente) levado em conta nos códigos de projeto.
Assim, o aço carbono e carbono-manganês (∼1,5%Mn), não obstante serem estruturados
por redes cristalinas CCC (cúbica de corpo centrado), são os primeiros a serem
considerados quando a faixa de temperaturas de trabalho vai de −45ºC até cerca de
450ºC. Exigências específicas que visam assegurar adequada tenacidade ao entalhe ou
tenacidade a fratura são requisitos adicionais exigidos nos serviços que envolverão
temperaturas de −45 até 0ºC. A resistência à fragilização pelo ″H2S úmido″ será
requerida a temperatura ambiente e pouco superiores, já a resistência à fluência
(>370°C), ao ataque promovido por misturas gasosas contendo H2S (>260°C), ao ácido
naftênico (>220°C), a oxidação (>530°C), entre outras, serão características requeridas à
temperaturas "elevadas". Tais requisitos, via de regra, acarretam a necessidade do
emprego de aços possuidores de propriedades específicas, por exemplo, aços ligados ao
Cr-Mo ou mesmo os aços inoxidáveis.
2.3.1. Soldabilidade
A adequada soldabilidade dos aços C e C-Mn, pode ser evidenciada pelo fato de que
para o serviço em ambiente não-fragilizante ou naqueles em que o requisito-tenacidade
não é exigido, dispensa-se a realização do pré e de pós-aquecimentos bem como do
controle da temperatura de interpasse na soldagem do aço que possua ″carbono
equivalente″ (CE), dado por:
CE = %C + %Mn
6+
(%Cr + %Mo + %V)
5+
(%Cu + %Ni)
15 (2.1)
e espessuras limitadas a 0,43/30mm, respectivamente [13]. Com relação ao tratamento
térmico para o alívio das tensões residuais (TTAT), que também tem o propósito de
promover o revenimento da ZTA, as ″regras″ diferem. Exemplificando, para o aço C/C-
Mn, o tratamento (595ºC mín.) é exigido apenas se a espessura da junta soldada for
superior a 38mm (1,5″). Sendo igual ou superior a 32mm mas inferior a 38mm, o

17 tratamento pode ser dispensado mediante a simples execução de pré-aquecimentos
que sejam superiores a 95ºC (200ºF). Tais condições trazem como conseqüências
imediatas a simplificação e o barateamento das operações de soldagem do aço carbono
(manutenção e fabricação) para os casos em que as restrições relativas ao CE, espessura,
tenacidade e fragililização não existirem. Por outro lado, aços liga com mais de 3%Cr
(isto é, aços com Cr entre 3 e 10%) deverão ser tratados em todos os casos; aqueles com
%Cr inferior, nem sempre. Ilustrando, um aço 2,25%Cr, deverá ser tratado (675ºC
mín.), apenas se a espessura for superior a 16mm (5/8″) [14]. Em todos os casos, evita-
se levar o aço à temperaturas intercríticas (entre as linhas A1 e A3 num diagrama de
equilíbrio Fe-C).
Há o interesse em analisar as razões pelas quais a espessura e o carbono equivalente de
um aço interferem tanto na sua soldabilidade. Para um aço com um dado CE, e para
uma solda realizada mediante um determinado aporte de energia ES, altas taxas de
resfriamento favorecerão a ocorrência de constituintes microestruturais pouco tenazes
ou pouco dúcteis em suas juntas soldadas. Daí, segue-se a necessidade do pré-
aquecimento e da manutenção de uma adequada temperatura de interpasse, que se
prestam para atenuar essa taxa. Peças espessas favorecem resfriamentos rápidos pelo
fato de que a dissipação de calor se faz, nestes casos, preponderantemente por condução
no sólido, ao passo que peças delgadas dissipam calor, principalmente, por radiação e
por convecção no ar através da superfície livre, usualmente mecanismos menos eficazes
de dissipação de calor. Por outro lado, para um dado aço, ES e espessura (vale dizer,
para uma mesma velocidade de resfriamento), maiores CE’s tenderão, tal como antes,
dar origem a constituintes com maior dureza e menos tenazes em suas ZTA′s pois, do
seu valor, têm-se uma indicação da temperabilidade do aço. [15, 16]
Em soldagem, a principal fonte de tensões residuais se origina das restrições existentes
à livre contração do metal de solda (MS) durante sua solidificação e resfriamento
subseqüente, restrição essa imposta pelas partes adjacentes menos aquecidas. Esse é um
fenômeno inevitável para os processos de soldagem que envolvem fusão e
aquecimentos localizados, sendo particularmente importante na soldagem de peças mais
espessas [15, 16].

18 De fato, fixado o CE, as restrições acima aludidas serão tanto mais severas quanto
maior for a espessura do componente. Tal quadro será ainda agravado se existirem
vínculos ou impedimentos externos originados de uma montagem excessivamente
rígida. Ainda assim, independentemente da existência de vínculos externos, à grandes
espessuras associam-se maiores volumes de solda e maiores níveis de restrições do que
o que seria produzido por peças menos espessas. Darão, assim, origem a campos mais
intensos de tensões residuais, os quais podem contribuir com a nucleação e o aumento
das taxas de propagação de trincas por fadiga em serviço, com o trincamento por
corrosão sob tensão, com a redução da resistência a fratura frágil da junta, e (contribuir)
de forma importante para a nucleação de defeitos de soldagem, tais como são a
decoesão lamelar ou a fissuração a frio. [15]
Na Figura 2.4 mostra-se, esquematicamente, a distribuição de tensões residuais numa
junta de topo, chanfro ″V″, em chapa com uma dada espessura. Nota-se que as tensões
longitudinais σL podem alcançar valores tão elevados quanto o limite de escoamento σy
do aço. Observa-se, ainda, que a inversão do sinal da tensão longitudinal (de tração para
compressão) dá-se a cerca de três larguras do cordão (L) relativamente à linha de centro,
o que indica haver superposição das tensões residuais de tração com regiões (ZTA) em
que microestruturas pouco dúcteis podem existir, fato que é particularmente grave. Tal
distribuição de tensões serve ainda para mostrar que numa chapa livre de vínculos
externos, o nível de restrição será sempre significativamente maior na direção paralela
ao cordão de solda do que nas direções perpendiculares, essas as geradoras da tensão
transversal σt. Dessa forma, não havendo suficiente rigidez, caso da soldagem em
chapas pouco espessas não vinculadas, as peças tendem a se deformar ou ″embicar″
mais fortemente na direção em que a restrição é menor, não obstante o carregamento
inferior.

19
Figura 2.4: Representação esquemática da distribuição de tensões residuais
longitudinais e transversais em um cordão de largura L de uma solda de
topo. [17]
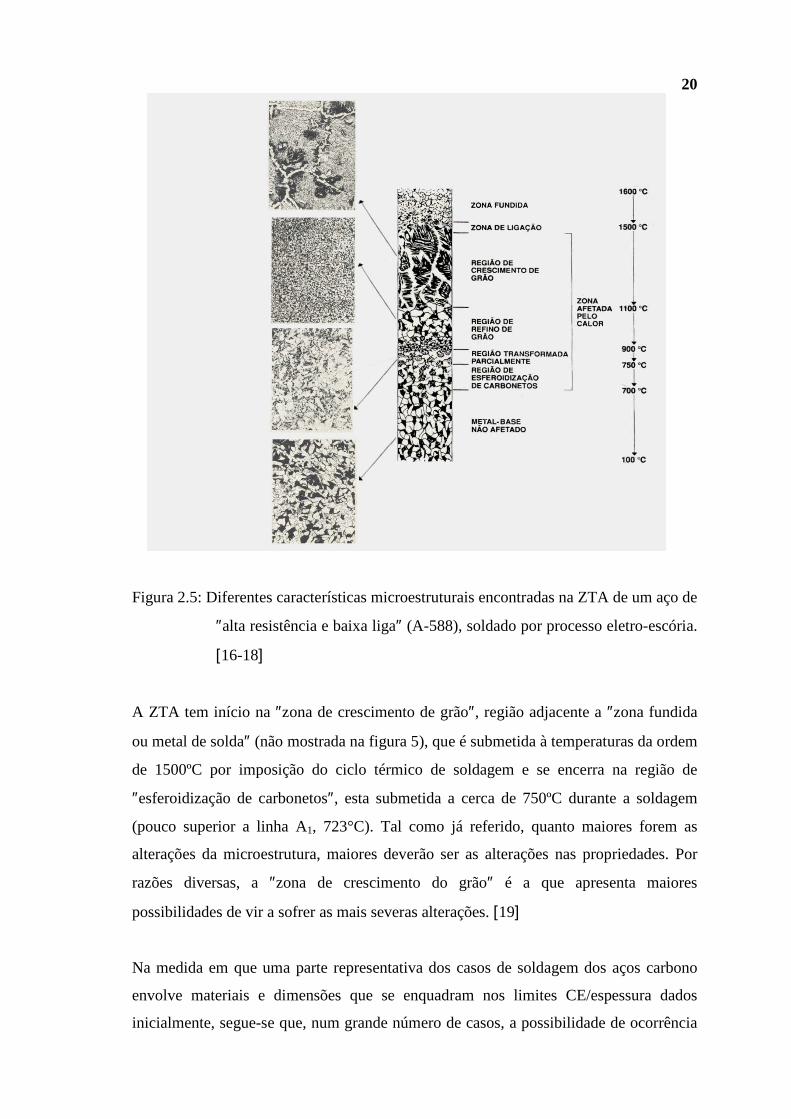
Sem entrar em maiores detalhes, a montagem apresentada na Figura 2.5 ilustra as
modificações microestruturais ocorridas a nível da ZTA e sofridas por um aço A-588,
aço com propriedades mecânicas e resistência à corrosão atmosférica superiores às do
aço carbono; baixa liga em Ni-Cr-Cu-V, devido a imposição de um ciclo térmico de
soldagem resultante da aplicação de um processo associado com altas energias de
soldagem, o processo ″eletro-escória″. Deve-se observar, em particular, a muito
significativa diferença entre a microestrutura da região de crescimento de grão com
aquela do metal base (MB). Tais diferenças são indicativas da possibilidade de se ter,
relativamente ao MB, importantes alterações de propriedades. Como já se fez menção, a
superposição de uma microestrutura pouco dúctil e tenaz, usualmente associada com
essa região, e altos níveis de tensões residuais de tração, a tornam particularmente
suscptível aos processos corrosivos e de fissuração [16, 17].
20
Figura 2.5: Diferentes características microestruturais encontradas na ZTA de um aço de
″alta resistência e baixa liga″ (A-588), soldado por processo eletro-escória.
[16-18]
A ZTA tem início na ″zona de crescimento de grão″, região adjacente a ″zona fundida
ou metal de solda″ (não mostrada na figura 5), que é submetida à temperaturas da ordem
de 1500ºC por imposição do ciclo térmico de soldagem e se encerra na região de
″esferoidização de carbonetos″, esta submetida a cerca de 750ºC durante a soldagem
(pouco superior a linha A1, 723°C). Tal como já referido, quanto maiores forem as
alterações da microestrutura, maiores deverão ser as alterações nas propriedades. Por
razões diversas, a ″zona de crescimento do grão″ é a que apresenta maiores
possibilidades de vir a sofrer as mais severas alterações. [19]
Na medida em que uma parte representativa dos casos de soldagem dos aços carbono
envolve materiais e dimensões que se enquadram nos limites CE/espessura dados
inicialmente, segue-se que, num grande número de casos, a possibilidade de ocorrência
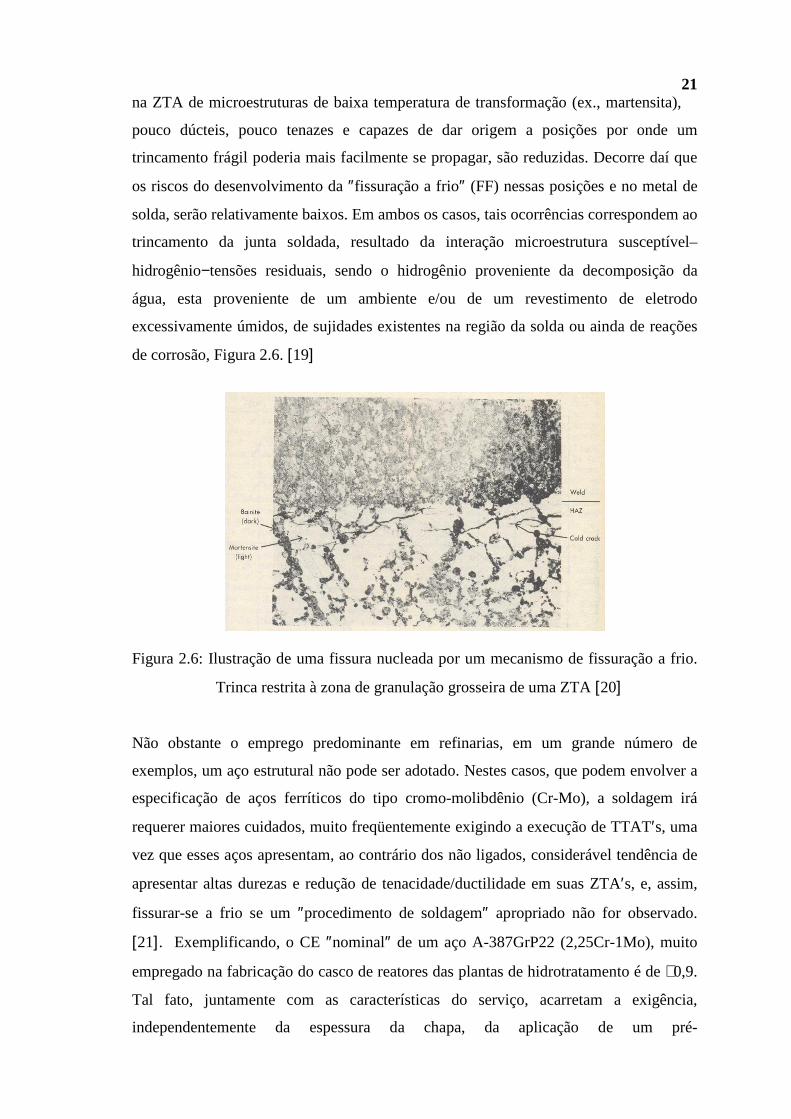
21 na ZTA de microestruturas de baixa temperatura de transformação (ex., martensita),
pouco dúcteis, pouco tenazes e capazes de dar origem a posições por onde um
trincamento frágil poderia mais facilmente se propagar, são reduzidas. Decorre daí que
os riscos do desenvolvimento da ″fissuração a frio″ (FF) nessas posições e no metal de
solda, serão relativamente baixos. Em ambos os casos, tais ocorrências correspondem ao
trincamento da junta soldada, resultado da interação microestrutura susceptível–
hidrogênio−tensões residuais, sendo o hidrogênio proveniente da decomposição da
água, esta proveniente de um ambiente e/ou de um revestimento de eletrodo
excessivamente úmidos, de sujidades existentes na região da solda ou ainda de reações
de corrosão, Figura 2.6. [19]
Figura 2.6: Ilustração de uma fissura nucleada por um mecanismo de fissuração a frio.
Trinca restrita à zona de granulação grosseira de uma ZTA [20]
Não obstante o emprego predominante em refinarias, em um grande número de
exemplos, um aço estrutural não pode ser adotado. Nestes casos, que podem envolver a
especificação de aços ferríticos do tipo cromo-molibdênio (Cr-Mo), a soldagem irá
requerer maiores cuidados, muito freqüentemente exigindo a execução de TTAT′s, uma
vez que esses aços apresentam, ao contrário dos não ligados, considerável tendência de
apresentar altas durezas e redução de tenacidade/ductilidade em suas ZTA′s, e, assim,
fissurar-se a frio se um ″procedimento de soldagem″ apropriado não for observado.
[21]. Exemplificando, o CE ″nominal″ de um aço A-387GrP22 (2,25Cr-1Mo), muito
empregado na fabricação do casco de reatores das plantas de hidrotratamento é de ∼0,9.
Tal fato, juntamente com as características do serviço, acarretam a exigência,
independentemente da espessura da chapa, da aplicação de um pré-

22 aquecimento/interpasse mínimos de 250ºC, juntamente com a aplicação do TTAT
referenciado anteriormente.
Os aços inoxidáveis formam uma terceira ″categoria″ de aços empregados em refinarias,
e entre eles, particularmente, os da série 300 ou austeníticos (estruturados por redes
cúbicas de face centrada, CFC), dos quais a referência é o aço 18Cr-8Ni, AISI 304.
Assim como a fissuração a frio se constitui num dos mais importantes defeitos a serem
evitados na soldagem dos aços ferríticos, a ″fissuração a quente″ (FQ) pode ser
considerada como sendo um dos principais defeitos (talvez pela freqüência com que
ocorrem) a serem evitados na soldagem dos aços inoxidáveis austeníticos. Tais trincas
podem nuclear-se no MS ou na ZTA. No MS, as também denominadas ″trincas de
solidificação″, dão-se devido a presença simultânea de estruturas resultantes da
solidificação do metal de solda, usualmente composta por grãos austeníticos dendríticos
ou celulares, de tensões residuais e de fases com baixo ponto de fusão, das quais o S e o
P tomam parte e cumprem importante papel, as quais são segregadas e permanecem no
estado líquido nos espaçamentos interdendríticos no curso da solidificação do MS,
Figura 2.7.a.
(a) (b)
Figura 2.7: (a) Trincas de solidificação nucleadas na superfície de um MS totalmente
austenítico que apresenta estrutura dendrítica [22] e, (b) Morfologia da
fase δ disposta em matriz austenítica. [23]
23 Dessa forma, a tendência à formação de FQ pode ser minimizada ao reduzir-se a
rigidez da montagem, observando-se uma rigorosa limpeza na região a soldar ajustando
nos níveis adequados (1 a 8%) a presença de uma ″fase δ″ na matriz austenítica do MS.
Para tanto, bastaria empregar o consumível adequado, AWS E-308 ou 309. Esta fase é
considerada benéfica por, entre outras razões, solubilizar as impurezas prejudiciais (S e
P) e dificultar a propagação de micro-fissuras eventualmente nucleadas por possuir
morfologia cuja geometria irregular desfavorece a propagação de trincas. Figura 2.7.b
[22, 23].
Adicionalmente às possibilidades do trincamento a quente, outros defeitos ou
dificuldades intrínsecas associadas à soldagem do aço inoxidável (os aços da série 400,
ferríticos e martensíticos, aí incluídos) devem ser consideradas. Todas essas
possibilidades, inclusive as trincas de solidificação, podem ser reunidas no diagrama de
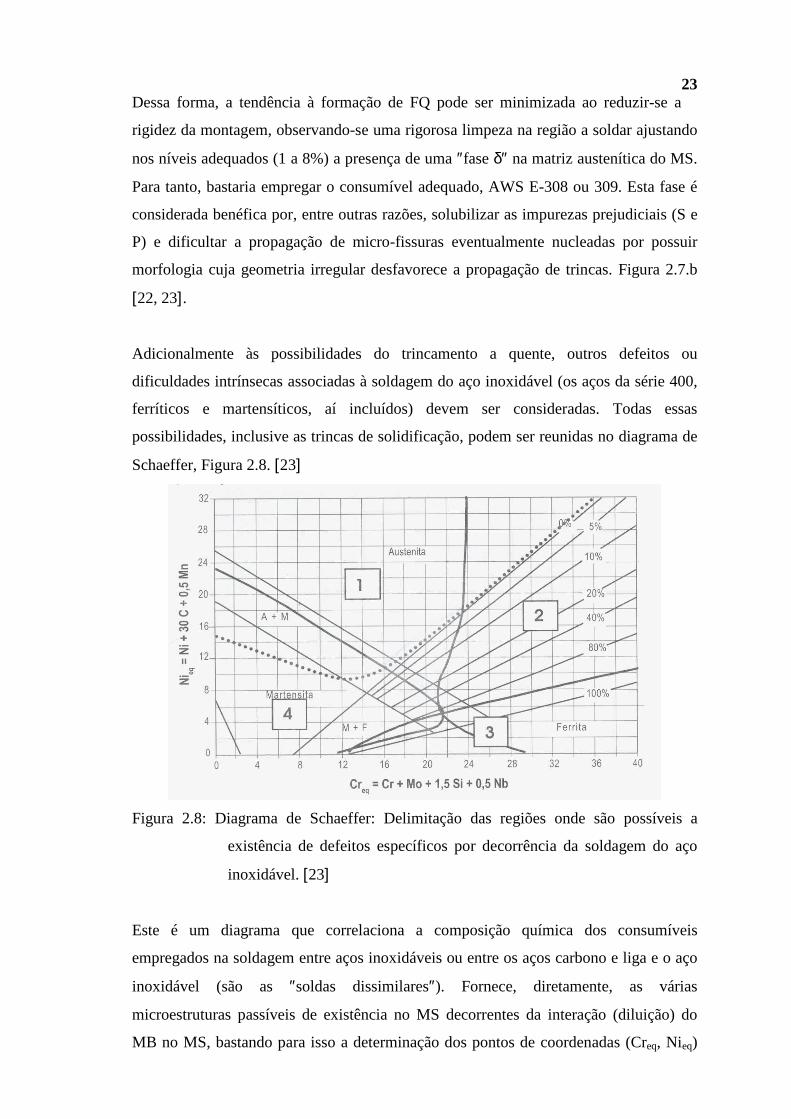
Schaeffer, Figura 2.8. [23]
Figura 2.8: Diagrama de Schaeffer: Delimitação das regiões onde são possíveis a
existência de defeitos específicos por decorrência da soldagem do aço
inoxidável. [23]
Este é um diagrama que correlaciona a composição química dos consumíveis
empregados na soldagem entre aços inoxidáveis ou entre os aços carbono e liga e o aço
inoxidável (são as ″soldas dissimilares″). Fornece, diretamente, as várias
microestruturas passíveis de existência no MS decorrentes da interação (diluição) do
MB no MS, bastando para isso a determinação dos pontos de coordenadas (Creq, Nieq)

24 associado a cada um deles, juntamente com o nível de diluição envolvido,
característico do processo de soldagem empregado. Encontram-se assinalados: campo 1
(austenítico), no qual é possível a fissuração a quente; campo 2 (austeno-ferrítico), no
qual é possível, mediante precipitação em serviço, a formação das fases fragilizantes σ e
α′; campo 3 (ferrítico), no qual é possível a fragilização da ZTA dos aços inoxidáveis
ferríticos devido ao crescimento excessivo do grão e campo 4 (martensítico), onde é
possível a fissuração a frio ou mesmo o trincamento provocado pelo contato de tais
regiões com o H2S em meio aquoso. O diagrama mostra ainda uma região central,
basicamente composta por austenita e ferrita (em torno do ponto (20, 10)), em que
nenhum dos mecanismos deve operar. [22, 23]
Entretanto, o diagrama de Schaeffer não fornece todas as informações. Em razão dos
ciclos térmicos e das tensões residuais associadas a uma soldagem, os aços da série 300
(mas não apenas) são susceptíveis de sofrer também o ataque intergranular no MB ou
ZTA, Figura 2.9, por ação de ácidos tal como são os politiônicos, H2SxO6, caso se
apresentem ″sensitizados″ e o trincamento por ″corrosão sob tensão″ por ação de
cloretos.
No caso da Figura 2.10, o condensado proveniente da injeção de vapor úmido, e uma
possível contaminação com cloretos, teria proporcionado, juntamente com tensões de
tração, a nucleação e a propagação da trinca, caracteristicamente transgranular.
Figura 2.9: Trincamento por corrosão sob tensão−intergranular ou IGSCC. Ácido
gerado a partir da interação ar (O2) +vapor (H2O) +sulfeto (S2−) sobre aço
AISI 304H sensitizado, material construtivo do fole de juntas de
expansão–UFCC; tubulação de interligação Regenerador-″Riser″. (Regap,
MG)

25
Figura 2.10: Trincamento por corrosão sob tensão−transgranular ou SCC em aço AISI
321. (Regap, MG)
Enquanto o ataque intergranular pode ser prevenido, utilizando-se, para isso, os aços
com baixo carbono, ≤0,03% (AISI 304L, 316L etc) e/ou a neutralização do ácido, o
trincamento por corrosão sob tensão transgranular irá requerer uma alteração mais
radical da metalurgia, como o uso de um SAF 2507, este um aço inoxidável ″austeno-
ferrítico″, ou ainda um AISI 904L, este um ″superaustenítico″. Pode ser realizada,
ainda, sendo factível, atenuação da agressividade do eletrólito, por exemplo, via redução
da temperatura (para valores inferiores a 60ºC), preferivelmente em paralelo com a
desaeração da solução.
São potencialmente sujeitos à falhas em serviço ou no ato da soldagem, as uniões
soldadas envolvendo materiais dissimilares. Exemplo freqüentemente encontrado
envolve a soldagem entre o aço carbono ou aço liga (ambos ferríticos) com o aço
inoxidável da série 300 (austenítico).
Além da possibilidade de formação do ″par galvânico″ e corrosão do metal menos
nobre, é também possível a nucleação de trincas por "fadiga térmica". Tal se daria
devido a ocorrência de flutuações das tensões térmicas σth originadas em uma junta
composta por materiais possuidores de diferentes coeficientes de dilatação, α,
submetidos a flutuações ou a variações de temperatura em serviço, incluindo-se paradas,
resfriamento, e partidas, aquecimento, de um dado equipamento, considerando-se a
existência de uma proporcionalidade que há entre uma e outra, σth∝α. Exemplificando,
à temperatura ambiente, enquanto o aço carbono ou aço liga possuem um coeficiente de
dilatação de cerca de 12 mm/mºC, o aço inoxidável austenítico possui coeficientes que
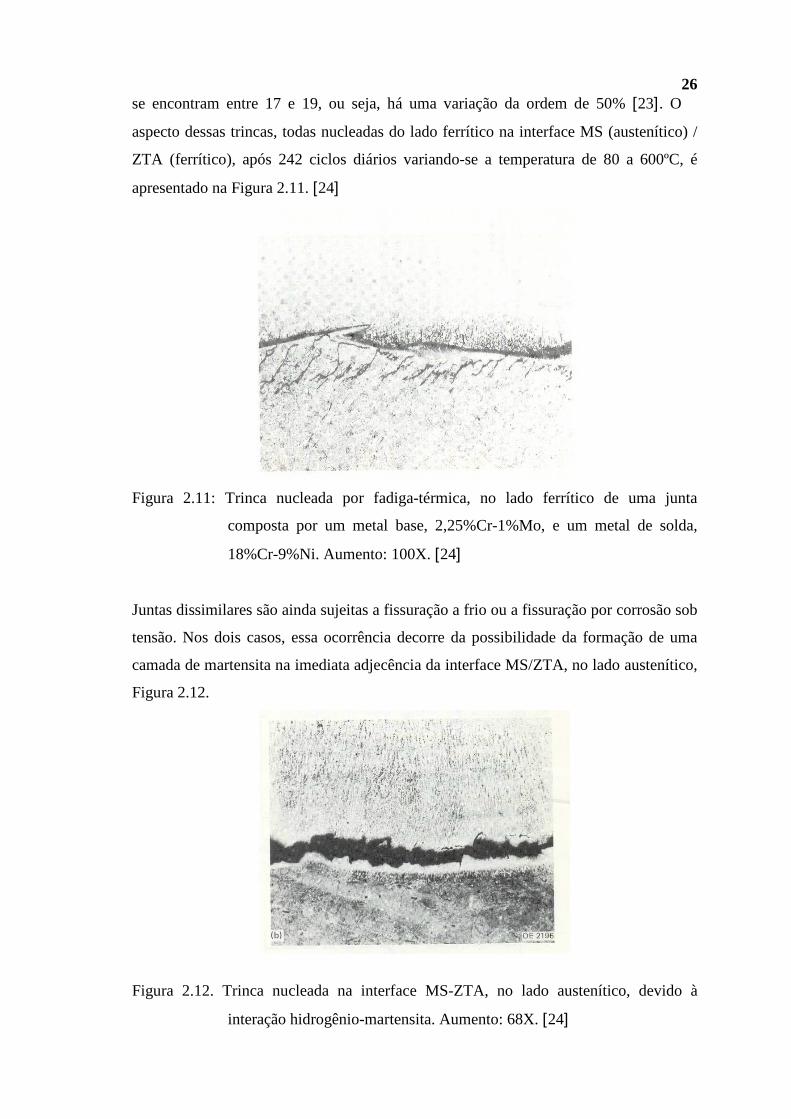
26 se encontram entre 17 e 19, ou seja, há uma variação da ordem de 50% [23]. O
aspecto dessas trincas, todas nucleadas do lado ferrítico na interface MS (austenítico) /
ZTA (ferrítico), após 242 ciclos diários variando-se a temperatura de 80 a 600ºC, é
apresentado na Figura 2.11. [24]
Figura 2.11: Trinca nucleada por fadiga-térmica, no lado ferrítico de uma junta
composta por um metal base, 2,25%Cr-1%Mo, e um metal de solda,
18%Cr-9%Ni. Aumento: 100X. [24]
Juntas dissimilares são ainda sujeitas a fissuração a frio ou a fissuração por corrosão sob
tensão. Nos dois casos, essa ocorrência decorre da possibilidade da formação de uma
camada de martensita na imediata adjecência da interface MS/ZTA, no lado austenítico,
Figura 2.12.
Figura 2.12. Trinca nucleada na interface MS-ZTA, no lado austenítico, devido à
interação hidrogênio-martensita. Aumento: 68X. [24]

27
Ambas as falhas podem ser prevenidas ou ter seu risco de ocorrência minimizado. A
interposição de uma ″almofada″ ou de uma camada possuidora de coeficiente de
dilatação intermediário ao do ferrítico e ao do austenítico (para tanto, é bastante usual
empregar-se ligas a base de níquel), se prestam para suavizar a transição e reduzir os
níveis das tensões térmicas, enquanto que a seleção do consumível adequado
(costumeiramente, um eletrodo AWS E-309), juntamente com o ajuste dos níveis de
diluição, esse diretamente associado com a energia de soldagem, serviriam para
prevenir a fissuração a frio [23, 24].
2.3.2 Corrosão em meio aquoso
Os aços carbono, bem como os aços ligados ao Cr-Mo, são materiais que, sob certas
condições, podem vir a apresentar importantes limitações relativamente a resistência a
corrosão em meio aquoso. Por meio aquoso, considera-se não apenas a imersão, mas
também o contato do aço com filmes condensados que, sob certas condições, se
formam e recobrem sua superfície. Diante dessas limitações, é comum encontrar-se o
aço protegido por um ″consórcio″ envolvendo esquemas de pintura e de proteção
catódica, como é o que comumente se faz com tubulação enterrada em solo agressivo
(úmido) e no fundo de tanques de armazenamento de petróleo (devido ao contato com a
salmoura que se separa do petróleo naquela região). Essa proteção pode também ser
implementada aplicando-se revestimentos metálicos resistentes ao meio na superfície
interna de vasos de pressão, tal como mostrado na Figura 2.13.

28 Figura 2.13: A foto ilustra a obtenção de revestimento interno (AISI 309L+AISI 347,
∼10mm total) por deposição de solda por eletro-escória em substrato de
aço Cr-Mo (A-387Gr22cl.2) no casco de um Reator HDT que
presentemente se encontra em operação. (Regap, MG)
Um processo corrosivo desenvolve-se de forma espontânea, no sentido termodinâmico
do termo. No entanto, em adição a esse aspecto, considere o que leva um bloco de aço
imerso em água a sofrer corrosão com base na seguinte analogia.
Quando curto-circuitado, o anodo (zinco) de uma ″pilha seca″ dissolve-se de forma
relativamente rápida num eletrólito formado por uma pasta úmida que contém o sal
NH4+Cl−, convertendo-se em cátions Zn2+ (Zn→Zn2++2e−), de modo a gerar corrente
elétrica. Estando a pilha desconectada ou em circuito-aberto, esse desgaste será muito
mais lento, mas ainda ocorrerá. Nessa condição, o lento descarregamento da pilha deve-
se à presença de traços de impurezas (por exemplo, ferro) na superfície do zinco em
contato com o eletrólito, os quais geram ″correntes de ação local″ oriundas de micro-
pilhas Zn-Fe curto-circuitadas, Figura 2.14. [25]
Figura 2.14: Representação esquemática de uma ″micro-pilha″ resultante da presença do
ferro como impureza no zinco, o anodo de uma pilha seca,
Fe2++Zn→Zn2++Fe. Inclui-se a corrente de corrosão Icorr, ou corrente de
ação local. A figura evidencia os quatro elementos necessários ao
estabelecimento de uma célula de corrosão. São eles: um condutor de
elétrons (zinco), um codutor de íons (pasta úmida contendo NH4+Cl−, o
eletrólito), um anodo (−) e um catodo (+)
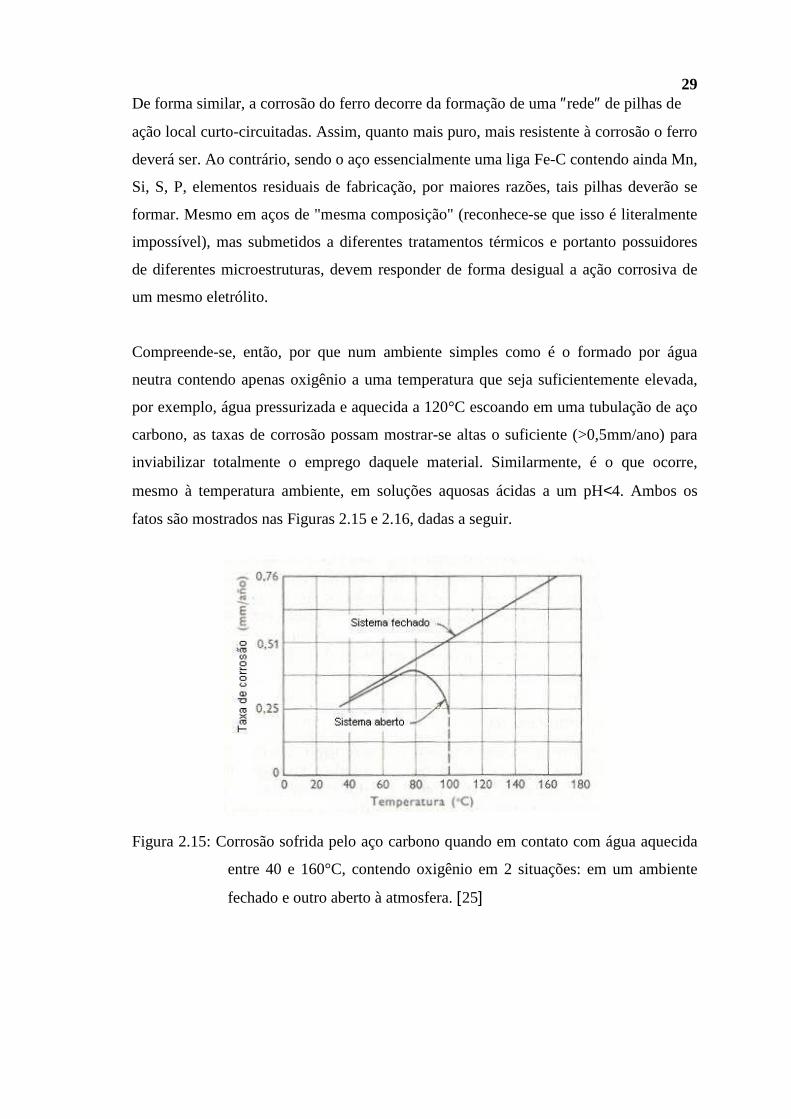
29 De forma similar, a corrosão do ferro decorre da formação de uma ″rede″ de pilhas de
ação local curto-circuitadas. Assim, quanto mais puro, mais resistente à corrosão o ferro
deverá ser. Ao contrário, sendo o aço essencialmente uma liga Fe-C contendo ainda Mn,
Si, S, P, elementos residuais de fabricação, por maiores razões, tais pilhas deverão se
formar. Mesmo em aços de "mesma composição" (reconhece-se que isso é literalmente
impossível), mas submetidos a diferentes tratamentos térmicos e portanto possuidores
de diferentes microestruturas, devem responder de forma desigual a ação corrosiva de
um mesmo eletrólito.
Compreende-se, então, por que num ambiente simples como é o formado por água
neutra contendo apenas oxigênio a uma temperatura que seja suficientemente elevada,
por exemplo, água pressurizada e aquecida a 120°C escoando em uma tubulação de aço
carbono, as taxas de corrosão possam mostrar-se altas o suficiente (>0,5mm/ano) para
inviabilizar totalmente o emprego daquele material. Similarmente, é o que ocorre,
mesmo à temperatura ambiente, em soluções aquosas ácidas a um pH<4. Ambos os
fatos são mostrados nas Figuras 2.15 e 2.16, dadas a seguir.
Figura 2.15: Corrosão sofrida pelo aço carbono quando em contato com água aquecida
entre 40 e 160°C, contendo oxigênio em 2 situações: em um ambiente
fechado e outro aberto à atmosfera. [25]

30
Figura 2.16: Corrosão sofrida pelo aço carbono em duas condições: em meio aquoso
alcalinizado pelo NaOH e acidificado pelo HC1, à temperatura ambiente.
[25]
A corrosão do ferro se passa com a liberação de um Faraday, F, (carga elétrica
equivalente a 1 mol de elétrons, ∼1,6×10−19C×6,0×1023≈96.500C/mol) por equivalente-
grama de ferro consumido no processo, ou seja:
1 F
eq -gFe =
96.500 A.s
28 gFe=
3446 A.s
gFe A = 2,9 x 10
gFe
s= 9145
gFe
ano mA 9,1
gFe
ano-4⇒ ⇒ ≅1 1 (2.2)
A unidade de ″mA″ empregada representa uma intensidade de corrente cuja ordem de
grandeza é mais compatível com aquela que se produz nos processos corrosivos [26].
Por exemplo, partindo-se de um bloco prismático de ferro (∼7,87g/cm3) com volume de
103cm3 (102 cm2 × 10 cm), e presumindo que a totalidade do processo corrosivo se
expresse por uma redução da altura ("espessura") do bloco, uma perda de 9,1 gFe
corresponderá a uma redução de cerca de 0,1 mm/ano, dimensão que permite de
imediato ter-se uma noção da severidade do processo corrosivo ou da agressividade do
eletrólito (a variação relativa correspondente à passagem 7870 g → (7870 − 9,1) ≅
7861g é de ∼ −0,1%. Então, a redução na altura será correspondente a 0,1% de 100
mm/ano).

31 Por sua vez, em meio neutro ou alcalino, o oxigênio viabiliza ou estimula, tal como
faz qualquer outro agente oxidante (por ex., Fe3+ ou Cr2O72− (≡ Cr6+)), a oxidação ou a
corrosão do aço:
Fe → Fe2+ + 2e− (2.3)
½ O2 + H2O + 2e− = 2OH−, (2.4)
o que acarreta a alcalinização do eletrólito na interface com o aço, o que vem favorecer
a precipitação do hidróxido:
Fe2+ + 2OH− → Fe(OH)2 (2.5)
Este, por sua vez, em meios pobres em oxigênio pode transformar-se em magnetita
(protetora) segundo a reação:
3 Fe(OH)2 → Fe3O4 + 2H2O + H2 (2.6)
É o que ocorre quando o aço faz contato com água para produção de vapor em caldeiras
(O2 ≤ 7 ppb). Entretanto, num ambiente aerado-úmido tal como o ar contendo uma dada
quantidade de vapor d’água, o hidróxido tenderá ser oxidado:
2 Fe(OH)2 + H2O + ½ O2 → 2 Fe(OH)3 → Fe2O3.H2O (−2H2O), (2.7)
gerando ″ferrugem″, este um óxido hidratado com capacidade protetora muito limitada,
o que vem justificar as sempre crescentes taxas de corrosão nestes ambientes (ou seja, o
aço não se passiva eficientemente nesses casos). Por outro lado, as taxas decrescem em
sistemas abertos por redução da solubilidade, e daí por redução da concentração do
oxigênio dissolvido a partir de cerca de 80ºC, como mostrado na Figura 2.15. Reduz-se,
assim, o poder oxidante, e dessa forma, a agressividade da solução. Nos sistemas
fechados tal não ocorre e as taxas de corrosão, que devem duplicar-se para cada
incremento de 30ºC na temperatura da água, dependerão apenas da eficácia do
transporte por difusão do oxigênio até a interface aço (sítios catódicos)−eletrólito
(Figura 2.15) [25, 27].

32
A corrosão sofrida pelo ferro em soluções alcalinas aeradas é esperada haja visto a
diferença entre os potenciais padrão de redução E0 do ferro e do oxigênio. Ambos são
medidos relativamente ao eletrodo de hidrogênio, cujo potencial, também nas condições
padrão, é arbitrado em 0mV. Esse eletrodo é formado por uma placa de platina, esta
apenas um suporte metálico com elevada inércia química, imerso em uma solução a
25°C com aH+=1→pH=0, em contato com gás H2 a 1 atm de pressão. No presente caso,
EFeo (–440mV) < EO
o
2 (+401mV), fato que leva o oxigênio a comportar-se como agente
oxidante (catodo) e o ferro como agente redutor (anodo) numa pilha Fe|Fe2+ || O2|OH−.
Trata-se de um processo favorecido termodinamicamente, ou seja, se passa com
decréscimo da energia livre de Gibbs, G, (∆G<0), a qual, para uma célula eletroquímica
à temperatura e pressão constantes, é dada por: [26]
∆ G = - n.F.EoCelo (2.8)
Assim, se a reação,
Fe + ½ O2 + H2O → Fe2+ + 2 OH−, (2.9)
deve se desenvolver espontaneamente da esquerda para a direita, o potencial da célula
Ecel tem que ser consistente com a equação 2.8, isto é, Ecel>0. De fato, sendo EOo
2> EFe
o ,
e E = E - E = E - E = 401- (-440) > 0Celo
redo
oxo
Oo
Feo
2, pode-se verificar que, ∆G0<0.
De forma semelhante, o menor potencial de redução do ferro frente ao do hidrogênio
(0mV), permite antecipar sua corrosão em meios ácidos não aerados. Neste caso, a
semi-reação de oxidação do ferro, Fe → Fe2+ + 2e−, também ocorre de forma que os
elétrons produzidos sejam consumidos na redução do próton, 2H+ + 2e− → 2Hads → H2,
ou, Fe + 2H+ → Fe2+ + H2↑, sendo a evolução do gás a etapa controladora do processo.
O oxigênio acentua significativamente a agressividade desses ambientes ao
proporcionar o aumento da velocidade de despolarização catódica ao reagir com o
hidrogênio, 2H + 1/2O2 → H2O. Como mostra a Figura 2.16, em meio ácido redutor
(HCl), taxas de 0,5mm/ano são alcançadas, ainda à temperatura ambiente, a um pH ≅ 3,
devendo duplicar para cada incremento de 10ºC na temperatura. Além da presença do

33 H+, que induz a oxidação do ferro ao reduzir-se, os meios ácidos são particularmente
agressivos pois ao manterem íons em solução, Fe2O3 + 6H+ + 2e− → 2 Fe2++3H2O,
impedem uma eventual passivação, expondo o metal direta e continuamente ao contato
com o eletrólito. [25]
A Figura 2.17 ilustra um caso em que uma desaeração ineficaz em água de caldeira
flamo-tubular aquecida a cerca de 100ºC (pH>7), proporcionou corrosão generalizada
da superfície externa dos tubos em aço carbono (A-178A), a uma taxa compatível com
aquela indicada no gráfico da Figura 2.15. Para essa condição, as taxas serão da ordem
de 0,5mm/ano. [28]
Figura 2.17: Aspecto externo da superfície corroída de um tubo (2″) A-178A de uma
caldeira flamo-tubular. Eletrólito: água aquecida a ∼100ºC a um pH>7
contendo oxigênio em excesso. Notar o furo e a corrosão preferencial
sofrida pela costura do tubo. (Regap, MG)
Além da corrosão generalizada, pode ser também observado processo corrosivo
intensificado na costura do tubo (soldagem por resistência elétrica), o que culminou
com a ocorrência de furo e a parada do equipamento. Por variadas razões, a região da
costura se apresenta mais susceptível à corrosão do que o metal base. Poder-se-ia
apontar como razões: tais regiões encontram-se mais tensionadas por efeito da presença
de tensões residuais, presumindo a não execução do TTAT; possuem uma
microestrutura ″bruta de fusão″, em princípio menos estável do que a do metal base que
lhe é adjacente. Adicionalmente, sendo a área anódica menor do que a catódica,
elevadas densidades de corrente acabam por impor severidade adicional. A qualquer

34 instante, no curso de um processo corrosivo que se processe por via eletroquímica,
devido ao requisito da manutenção da neutralidade elétrica ou de conservação da carga,
deve cumprir-se rigorosamente: ∑iA = ∑iC, sendo ∑iA o somatório das correntes
anódicas e ∑iC o somatório das correntes catódicas. Assim, as densidades de corrente
deverão atender à desigualdade, ∑iA/AA >> ∑iC/AC, pois AA << AC. [26]
Além de promover processos corrosivos cuja ação leva à corrosão uniforme, o oxigênio
pode também dar origem a ″pilhas de aeração diferencial″. Na medida em que toma
parte de semi-reações catódicas, posições adjacentes em contato com água com
diferentes níveis de aeração deverão (uma delas) se comportar anodicamente ou se
corroer. Mais especificamente, aquela em contato com ambiente mais aerado tenderá a
se comportar como um catodo e a adjacente como um anodo. Depósitos porosos ou
tubérculos que se formam sobre a superfície interna de tubulações que conduzem água
aerada contendo bactérias aeróbicas que se alimentam de oxigênio, favorecem muito
particularmente esse tipo de ataque. Figura 2.18. [27]
Figura 2.18: Corrosão por aeração diferencial decorrente da formação de tubérculos em
tubo API 5L-B condutor de água para o combate a incêndio. Notar,
mais uma vez, o ataque preferencial ao cordão de solda. (Regap, MG)
Dada a posição que o aço carbono ocupa na ″série galvânica″, cuidados específicos
devem ser observados relativamente à possibilidade do desenvolvimento da corrosão
por ″par galvânico″, em que o aço possa se comportar como anodo. É, por exemplo, o

35 que ocorre em soldas dissimilares envolvendo o aço carbono e os aços inoxidáveis.
Na foto dada a seguir, Figura 2.19, mostra-se a corrosão severa que incidiu em um
flange em aço carbono, A-234GrWPB, devido a formação de par galvânico com tubo
em aço inoxidável, A-312Gr304, quando todo o conjunto se encontrava imerso em uma
solução ácida contendo oxigênio (devido ao contato com o ar) e sulfato de alumínio,
agente floculante empregado no tratamento de água.
Figura 2.19: Corrosão por par galvânico de um flange em aço carbono (A-234GrWPB)
devido ao contato (solda) com tubo em aço inoxidável (A-312Gr304),
estando ambos imersos em solução aquosa aerada contendo sulfato de
alumínio. (Regap, MG)
Este sal, ao hidrolizar-se, pode dar origem a eletrólitos ácidos altamente agressivos,
como evidencia a reação a seguir,
Al 2(SO4)3 + 6 H2O → 2 Al(OH)3 + 3 SO32− + 6 H+, (2.10)
observando-se que o sentido de progressão da corrosão dá-se do centro (interface com a
parede do tubo) para as bordas do flange.
A agressividade de um ambiente desaerado contendo ácido carbônico (∼100ºC) ao aço
carbono, é evidenciada a seguir. Figura 2.20.

36
Figura 2.20: Ataque ácido (H2CO3) em ambiente não aerado que incidiu internamente
no casco em aço carbono (A-515Gr60) de um trocador de calor
″amina-amina″ de uma unidade de extração de CO2 de uma corrente
H2+CO2. (Regap, MG)
A semi-reação (catódica) correspondente foi,
H2CO3 + e− → HCO3− + H, (2.11)
deixando, neste caso, de envolver diretamente o H+ por tratar-se de um ácido fraco que,
em princípio, deveria ser pouco agressivo, e como semi-reação anódica,
Fe → Fe2+ + 2 e−, (2.12)
com a reação global,
Fe + H2CO3 → FeCO3 + H2, (2.13)
podendo precipitar-se o carbonato, não necessariamente protetor. As condições sob as
quais uma passivação deverá ser efetiva poderão ser inferidas a partir da determinação
dos valores da taxa de corrosão (Vcor, mm/ano) no aço carbono mediante emprego da
relação de Waard-Millians:

37
log (V ) = 5,8-1719
273,15+ t+ 0,67 log (P )cor CO2
, (2.14)
onde t é a temperatura (ºC) e PCO2 é a pressão parcial de CO2 (bar). [29,30]
Nos meios ácidos aerados, a semi-reação catódica prevalecente é indicada pelo
equilíbrio,
O2 + 4 H+ + 4 e− = 2 H2O, Eº = +1229 mV, (2.15)
observando-se, mais uma vez, que na presença do oxigênio a polarização do catodo via
adsorção do hidrogênio (H+ + e− → Hads) é desfavorecida, fato que serve para acentuar
ainda mais a agressividade desses eletrólitos. Notar, também, que a semi-reação acima
origina uma diferença de potenciais, relativamente ao potencial de redução do ferro,
ainda maior do que nos casos vistos anteriormente.
A seguir, Figura 2.21, tem-se um exemplo do ataque levado a efeito pelo ácido sulfúrico
em meio aerado (ar). O ácido teria sido gerado em serviço através da interação das
cinzas produzidas pela queima de óleo combustível rico em compostos sulfurados com
condensado proveniente de vazamento pelo bloqueio (válvula gaveta) de um tubo
ramonador, tubo destinado a injetar vapor para manter limpa a superfície externa desses
tubos em operação. Na Figura 2.21, nota-se a ocorrência de dois furos na região com
alvéolos e o aspecto liso em parte da região corroída, esta em nível suficiente para
acarretar o rasgamento longitudinal por efeito das tensões circunferenciais que se
originam da pressurização interna do tubo. [31]

38
Figura 2.21: Corrosão externa promovida pelo H2SO4 diluído e aerado em tubo 2″, A-
178A, de caldeira aquo-tubular. (Regap, MG)
Considerando que os sulfatos usualmente se fazem presentes nas cinzas, poder-se-ia ter:
Al 2(SO4)3 + 3 H2O → 3 H2SO4 + Al2O3. (2.16)
Diluído, esse ácido apresenta um grau de ionização de 61%, H2SO4→H++HSO4−,
podendo o H+ tomar parte da semi-reação de redução mais uma vez às custas da
oxidação do ferro. Não diluído, o ácido tem características oxidantes, o que termina por
favorecer a passivação do aço pela formação e precipitação do sulfato, FeSO4, insolúvel
na solução concentrada, viabilizando o emprego do aço carbono para, por exemplo, o
armazenamento do ácido sulfúrico concentrado, ∼70 a 99% [27, 32]. Aparentemente,
soluções aquosas com concentrações inferiores a cerca de 70% já seriam suficientes
para hidrolizar o sulfato e despassivar o aço, o que deveria ocorrer segundo a seguinte
reação:
FeSO4 + 2 H2O → Fe(OH)2 + H2SO4 (2.17)
Além do oxigênio, o cloreto normalmente se faz presente nas águas empregadas
industrialmente em refinarias. É o caso da "água de refrigeração", usada como fluido
refrigerante em um grande número de permutadores-resfriadores ou condensadores no
processo, não obstante sofrer tratamento para ajuste da incrustrabilidade e da
corrosividade, tendo em vista essa aplicação. De fato, ao menos em parte, o cloreto deve

39 decorrer da injeção de compostos tais como o CaClO2, feita com o objetivo de inibir
o crescimento biológico.
O diagrama de Pourbaix para o sistema Fe×H2O−25ºC, Figura 2.22, ilustra o efeito do
cloreto na corrosão do aço quando o mesmo se apresenta em várias concentrações na
água à temperatura ambiente, 10−3, 10−2, 10−1 e 100 íon-g/L. A sua presença pode
acarretar uma significativa redução do domínio de passivação do aço ou aumento do
domínio onde o Fe2+ é estável.[33].
Figura 2.22. Diagrama de Pourbaix para o Fe-H2O-25ºC. Mostra-se o efeito do cloreto
na passivação do ferro em várias concentrações. [33]
Tais diagramas reúnem em domínios, o equilíbrio, dado em função do potencial de
eletrodo e do pH medidos na interface metal-eletrólito, que há entre o metal no estado
reduzido (imune a corrosão neste estado), ionizado (corroído) e como óxido, neste caso
podendo passivar-se de forma efetiva a depender da presença ou não de contaminantes
agressivos no eletrólito (o cloreto é um deles), das características do filme quanto a
aderência, porosidade e a resistência que oferece ao transporte de íons e elétrons [33].
No presente exemplo, concentrações crescentes de cloreto reduzem progressivamente o
domínio em que o aço poderia ser protegido ao passivar-se. Em particular, quando em

40 contato com água contendo 3550 ppm Cl− (100 íon-g/L), a solução, mesmo com pH
próximo a 9, ainda leva à desestabilização do filme passivado, expondo localmente o
metal ao eletrólito. Formam-se, então, pilhas na qual a área anódica geralmente tem
dimensões inferiores às da área catódica, dando início assim à corrosão localizada ou
″alveolar″ nos aços, em contraposição à corrosão uniforme. Nessas áreas, ou nas
cavidades a elas associadas, onde, por qualquer razão, a renovação do eletrólito é difícil,
a solução ali retida tende a se acidificar devido aos efeitos de hidrólise dos produtos de
corrosão, o que se dá segundo a reação,
FeCl2 + 2 H2O → Fe(OH)2 + 2 H+Cl−. (2.18)
Exemplificando, uma solução 1N-FeCl2 possui um pH=2,1 à temperatura ambiente. [27,
33].
A perda localizada da passivação assume particular importância quando estão
envolvidas as ligas passiváveis ou as ligas ″ativo-passivas″, pois desta perda pode-se ter
desenvolvido qualquer um dos fenômenos: o pite, a corrosão em frestas (″crevice″) ou o
trincamento por corrosão sob tensão. Como exemplo de ligas passiváveis, tem-se os
aços inoxidáveis ou o alumínio e suas ligas. Por sua vez, dada a limitada capacidade de
proteção conferida pelos óxidos, sulfetos, sulfatos ou carbonatos de ferro gerados em
meio aquoso, o aço carbono, ou mesmo os aços liga Cr-Mo, não podem ser incluídos
nesta classificação.
A formação do filme protetor em uma liga ativo-passiva em meio aquoso aerado, deve
iniciar-se pela oxidação do elemento mais reativo, M → M2+ + 2e−, o que se passa em
paralelo com a redução do oxigênio (equação 2.4). Segue-se a formação de um
complexo intermediário, M2+ + OH− → M(OH)+, que ao ser complexado por moléculas
de água dará origem a um filme com estrutura similar à de um gel (flexível e rico em
moléculas de água) que tende a recobrir e aderir ao metal liberando H+ no processo.
Uma vez formado, a (lenta) dissolução do filme é compensada pelo transporte dos íons
M2+ e/ou O2− via lacunas catiônicas/aniônicas, até que se atinja um ″estado estacionário″
em que a espessura do filme se mantém essencialmente constante, usualmente <10nm
(<10−5mm). Na Figura 2.23, é mostrada a representação esquemática da estrutura do

41 filme desenvolvido sobre os aços inoxidáveis num estágio inicial e após sua
formação. Figuras 2.23 (a) e (b). [34]
Figura 2.23: (a) Modelo representativo de um filme em desenvolvimento em uma liga
ativo-passiva em sua parte central e, (b) com o filme já formado. Notar a
abundância de moléculas de água no filme [34]
Entre os defeitos existentes no substrato e em sua superfície, as quais podem afetar ou
perturbar a continuidade do filme, encontram-se as inclusões não metálicas, sempre
presentes nos aços [34]. Tais perturbações enfraquecem localmente a camada passivada
e podem dar origem a corrosão por pites. Este é um fenômeno que, diferentemente da
corrosão uniforme em que os sítios anódicos-catódicos devem continuamente se
intercambiar, a corrosão por pites é extremamente localizada, vindo, necessariamente,
do resultado de uma clara definição dos sítios anódicos.
Então, dada à muito desfavorável relação entre as áreas anódicas e catódicas nestes
casos, as densidades de corrente anódicas serão usualmente muito altas e, como
resultado, forma-se um pite cuja característica é possuir uma elevada relação
profundidade/diâmetro, como vistos nas Figuras 2.24 e 2.25. Além de possibilitarem
falhas por vazamentos, atuam também como ″eficazes″ concentradores de tensão, sendo

42 relativamente comum encontrar-se nucleadas, a partir de sua extremidade, trincas
resultantes de um processo de corrosão sob tensão.
Figura 2.24: Ilustração da grande incidência de pites na válvula (AISI 316, ∅ 5cm) de
uma bandeja posicionada na parte superior de uma torre extratora de H2S
do diesel de uma unidade de hidrotratamento. Carga (provavelmente)
contaminada com cloretos. (Regap, MG)
Figura 2.25: Ocorrência de pites na superfície interna de um tubo (∅ 3/4″-2,1 mm de
parede) em A-213Tp321 de um trocador de calor que operava com o
efluente do reator de uma planta de hidrodessulfurização, fluido em que
não se espera uma presença importante de cloretos. Notar a ausência de
desgaste por corrosão na parede do tubo. (Regap, MG)

43 Foi afirmado que os cloretos podem danificar filmes originalmente perfeitos.
Considerando as ligas passiváveis, e de acordo com um dos mecanismos propostos, o
cátion M2+ se combinaria com o cloreto para formar complexos que se solubilizariam no
eletrólito (como MCl+(H2O)). Nessas posições, ânions (cloreto) substituiriam a água e o
O2−, promovendo, assim, uma completa descaracterização do filme original naquela
posição. Por sua vez, a destruição local do filme possibilitaria uma elevada produção de
cátions devido ao contato do metal desprotegido com o eletrólito, o que acarretaria o
ingresso de mais ânions de forma a restabelecer a neutralidade de cargas. A presença
conjunta de cátions, cloretos e água conduz à formação do H+Cl− por hidrólise do sal,
acidificando a região, impedindo a repassivação, e viabilizando a formação do pite. Esse
mecanismo é esquematizado a seguir, na Figura 2.26. [34]
Figura 2.26: Esquema ilustrativo da formação de um pite. [34]
Meios fortemente alcalinos ou cáusticos (por exemplo, meios aquosos contendo uma
base forte tal como o NaOH em concentrações superiores a 5%), podem também
ocasionar a corrosão do aço que, nestas condições, se comporta como um metal
anfótero. O ataque ao aço pode ser representado pelas reações,
Fe + 2 NaOH → Na2FeO2 + 2 H, ou, (2.19)
Fe → Fe2+, (2.20)

44 formando-se, como produto de corrosão, o ferrito de sódio FeO2
2− que é solúvel no
eletrólito. Figura 2.27. Paralelamente, produz-se hidrogênio (tal como em meio ácido-
redutor), o qual pode migrar ou difundir-se no aço e, neste caso, podendo produzir-se a
″fragilização cáustica″, que se desenvolve sob a forma de trincas intergranulares. [27,
35]
Figura 2.27: Processo corrosivo sofrido pelas aletas de um tubo de aço carbono de um
permutador de calor que em operação aquecia uma solução cáustica a
50ºC. (Regap, MG)
2.3.3 Corrosão no aço trabalhado a frio, corrosão-erosão, corrosão-fadiga, cavitação
e o trincamento assistido por tensão. A sensitização do aço inoxidável
O aço carbono trabalhado ou deformado a frio, aquele deformado à temperaturas nas
quais um processo de formação de novos grãos com baixas densidades de defeitos não
ocorra, isto é, não ocorra a recristalização do aço, experimenta maiores taxas de
corrosão do que um outro a ele equivalente mas não encruado, quando em contato com
meios ácidos. A precipitação de carbonetos ou de nitretos, ou ainda, a formação de uma
″atmosfera″ de átomos de carbono e de nitrogênio em pontos da rede cristalina do aço
em que se verifique um acúmulo de imperfeições produzidas pela deformação
responderia por essa maior reatividade. Tal se daria pela redução da sobretensão ao
hidrogênio nessas posições relativamente à matriz ferrítica adjacente (portanto, em meio
aquoso neutro essa maior reatividade não deveria se verificar), ou seja, um aço encruado
tem reduzida a energia de ativação associada à reação de redução do hidrogênio ou de
despolarização catódica, [25],

45
H+ + e− → ½ H2. (2.21)
Além dos processos ″puramente″ eletroquímicos nos quais o encruamento do aço
intervêm, a corrosão do aço pode ser assistida por componente mecânico externo, que
pode ou não predominar sobre o eletroquímico. Esse processo é denominado corrosão-
erosão.
Um processo de corrosão-erosão é caracterizado por envolver, seguidamente, 1) a
corrosão (componente eletroquímico), e a geração de um “produto de corrosão” que se
precipita recobrindo e passivando o metal, seguido de, 2) sua remoção por erosão
(componente mecânico), expondo o metal, agora desprotegido, a um novo ataque
corrosivo (bem como erosivo!). [26]
Tal seqüência de eventos pode desenvolver-se sempre que houver movimento relativo
entre um eletrólito e uma superfície metálica. Por conseguinte, é fácil depreender que
pode operar em inúmeras situações, devendo responder por um significativo número de
casos dados como tendo sido apenas corrosão.
São particularmente susceptíveis os metais macios tais como o cobre e ligas, o alumínio
e ligas, e outros similares, mas, de um modo geral, todas as ligas, incluindo os aços nas
suas mais variadas formulações e durezas, poderão sofrer esse tipo de dano desde que se
façam presente as condições apropriadas [26]. Do mesmo modo, serão particularmente
susceptíveis os acessórios de tubulações que promovam turbulência localizada (como
nos internos de válvulas ou bombas, ou ainda em reduções ou trecho reto de tubo que
vem imediatamente a jusante), ou que interfiram e modifiquem a direção do fluxo.
Serve para caracterizar essa influência, o fato de que nenhuma erosão foi constatada na
redução, de mesma metalurgia e geometria à mostrada na Figura 2.28, que se
posicionava a montante de uma válvula controladora de vazão.

46
Figura 2.28: Corrosão-erosão interna (com provável predominância de erosão) em
redução (A-234GrWPB) posicionada à jusante de uma válvula
controladora de vazão de carga do reator de uma unidade de
hidrotratamento (diesel+H2+particulado em suspensão+água) a cerca de
200ºC-45kgf/cm2.man. (Regap, MG)
Em princípio, a corrosão–erosão pode se dar tanto em meio “monofásico”; por exemplo,
água acidificada com CO2 sem segundas fases sólidas ou bôlhas de qualquer natureza,
ou em meio “bifásico”, servindo para exemplificar, água acidificada com CO2 e FeCO3
como particulado sólido em suspensão, este o produto de corrosão desagregado e
fragmentado. No primeiro caso, o responsável pela remoção do filme protetor serão
unicamente as tensões cisalhantes geradas na interface parede-meio, e no segundo, além
das tensões, o impacto causado pelas partículas sólidas. Dado os tipicamente reduzidos
valores das tensões, por exemplo, a 66m/s com grau de vaporização zero, um gasóleo
efluente de uma destilação a vácuo a 370°C, com densidade de 0,7 e viscosidade
cinemática de 5×10−7m2/s, gerou uma tensão cisalhante de apenas 10,3kPa ou
∼0,1kgf/cm2 [36], a severidade do desgaste causado pelos processos de corrosão-erosão
em escoamentos bifásicos serão, comumente, mais elevados do que aquele causado
unicamente por escoamentos monofásicos, para um mesmo número de Reynolds.
Entretanto, o papel da tensão cisalhante em quaisquer dos casos não pode ser
subestimado. As tensões cisalhantes são as indutoras de turbulência na imediata
adjacência das superfícies metálicas, afetando fortemente a taxa de transferência do
composto agressivo da solução até a interface meio-metal [37]. Some-se a isto, a usual

47 aderência deficiente dos filmes superficiais produzidos no aço carbono e liga, por
exemplo, sulfetos e carbonatos. Então, nestes casos, é possível esperar que tais produtos
de corrosão se convertam rapidamente em agentes erosivos ao passarem a incorporar a
solução.
Forças (Fc) localizadas, muito intensas, normalmente resultam do colapso de bolhas de
vapor nucleadas sobre superfícies metálicas. Se essa força superar a associada ao limite
elástico do metal, Fc>σyA, sendo A a área sob a qual a força Fc atua, pode ocorrer a
deformação da superfície e, eventualmente, o próprio arrancamento de partículas
metálicas pode se dar. Tal processo é denominado ″cavitação″. [26]. Na Figura 2.29,
ilustra-se o intenso desgaste promovido por cavitação em um tubo de aço liga. Trata-se
de uma derivação destinada a injetar vapor de média pressão em linha de resíduo
(operação intermitente) numa unidade de coqueamento retardado. A configuração
adotada estaria proporcionando a entrada de hidrocarboneto e de bolhas na posição, ao
que se seguiria o colapso dessas últimas.
Figura 2.29: Desgaste produzido por cavitação em derivação de 2″, A-335GrP5 (5%Cr-
0,5%Mo). (Regap, MG)
Além dos processos de desgaste mecanicamente assistidos vistos, o trincamento
intergranular ou transgranular das ligas metálicas via participação de carregamentos
cíclicos em meios corrosivos, denominado ″corrosão-fadiga″, ou não-cíclicos, residuais
ou aplicados, denominado ″corrosão sob tensão″ (SCC e IGSCC, já mencionados), são
da mais alta relevância pelo número de casos de falhas que tais mecanismos se
encontram envolvidos. Verifica-se que as juntas soldadas não aliviadas do aço carbono

48 podem sofrer o trincamento quando em contato com soluções alcalinas a um pH
9∼11, contendo CO2, NH3 e S2−, tal como ocorre nos sistemas de topo de torres
fracionadoras, com soluções cáusticas ou com nitratos (mas não nitretos), e ainda com
amônia anidra+ar. Encontra-se também sujeito à corrosão sob tensão o aço inoxidável
da série 300 quando em contato com soluções orgânicas ou não contendo cloretos e
brometos. Se sensitizado, estará sujeito ao trincamento intergranular, quando em contato
com soluções contendo ácidos politiônicos, cloretos e ainda água+oxigênio a alta
temperatura [25, 26, 32].
Como exemplo do trincamento intergranular produzido por corrosão-fadiga, segue o
(trincamento) sofrido por um componente (corpo) em aço AISI 304H (0,04−0,10C) de
uma junta de expansão que em serviço fazia contínuo contato com vapor úmido,
possivelmente contaminado com cloretos (vapor com ∼17kgf/cm2 em temperatura
inferior a de saturação, ∼200ºC). Embora em operação estivesse protegido por uma
″camisa″ interna do contato direto com gases e com catalisador, esses à temperaturas
próximas a 750°C, o aço 304H é, em operação, certamente aquecido a um nível
suficiente para dar origem a sucessivos ciclos de secagem (evaporação) após seu
molhamento ou umidecimento. Nessas circunstâncias, a superfície ou região
inicialmente umedecida tende a se contrair enquanto o metal mais abaixo restringe essa
tendência, o que termina por produzir tensionamentos que são relaxados após a
evaporação do líquido retido na superfície (a superfície retoma sua temperatura inicial).
Tais ciclos teriam promovido seguidamente o tensionamento e a relaxação dessas
tensões o que, juntamente com uma microestrutura ″sensitizada″ (ZTA e MB), foi capaz
de ocasionar o trincamento intergranular do aço. Nota-se nas Figuras 2.30 (a) e (b), que
o trincamento se restringe à ZTA e ao MB, não atingindo o cordão, fato que serve para
mostrar o papel da microestrutura, e, em particular, dos contornos de grãos, neste
fenômeno.

49
(a)
(b)
Figura 2.30: (a) Vista de topo de uma amostra trincada por corrosão-fadiga. Amostra
extraída do corpo soldado (1/2″ de espessura - AISI 304H) de uma junta
de expansão de uma UFCC, posição que em serviço fazia constante
contato com vapor úmido e, (b) Evidência de uma propagação
intergranular. (Regap, MG).
Não obstante a tendência que os aços inoxidáveis têm de sofrer o trincamento
trangranular em meios contendo cloretos, o comprometimento dos contornos de grão
relativamente à resistência a corrosão original da liga foi, então, suficiente para produzir
ou potencializar uma trajetória de propagação da trinca, o que se faria mediante a
dissolução preferencial desses contornos, sendo o papel das tensões alternadas o de
acelerar essa dissolução.
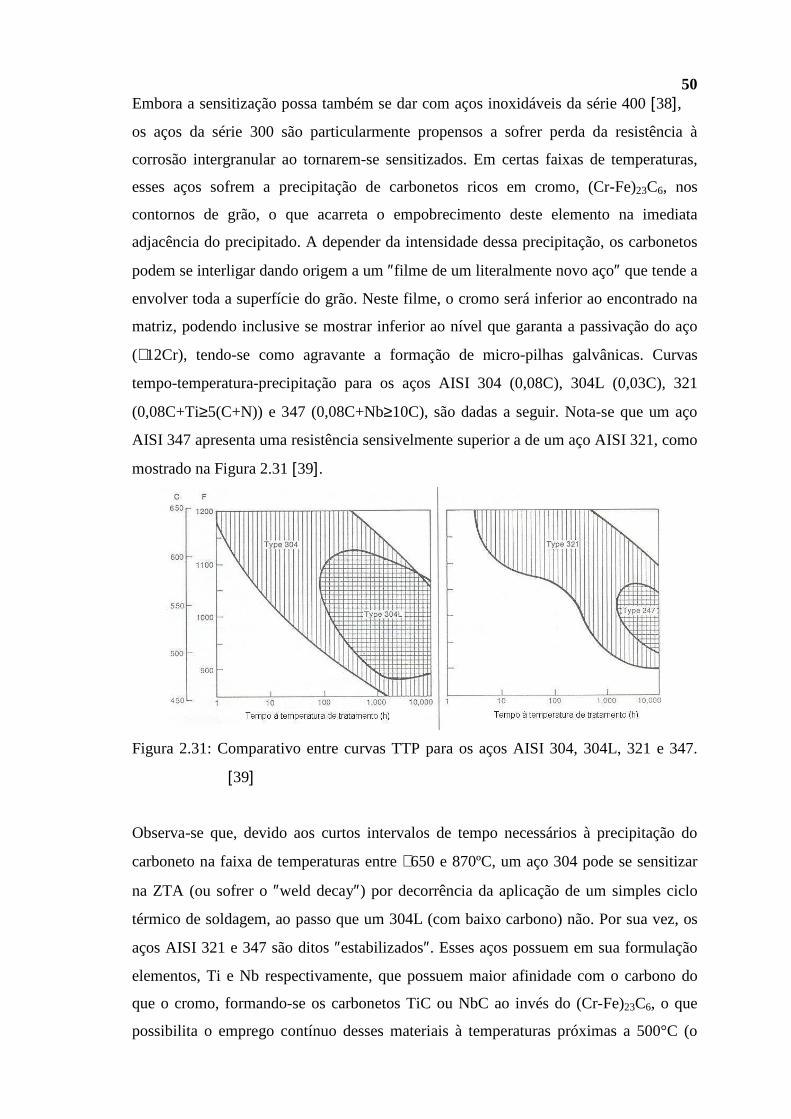
50 Embora a sensitização possa também se dar com aços inoxidáveis da série 400 [38],
os aços da série 300 são particularmente propensos a sofrer perda da resistência à
corrosão intergranular ao tornarem-se sensitizados. Em certas faixas de temperaturas,
esses aços sofrem a precipitação de carbonetos ricos em cromo, (Cr-Fe)23C6, nos
contornos de grão, o que acarreta o empobrecimento deste elemento na imediata
adjacência do precipitado. A depender da intensidade dessa precipitação, os carbonetos
podem se interligar dando origem a um ″filme de um literalmente novo aço″ que tende a
envolver toda a superfície do grão. Neste filme, o cromo será inferior ao encontrado na
matriz, podendo inclusive se mostrar inferior ao nível que garanta a passivação do aço
(∼12Cr), tendo-se como agravante a formação de micro-pilhas galvânicas. Curvas
tempo-temperatura-precipitação para os aços AISI 304 (0,08C), 304L (0,03C), 321
(0,08C+Ti≥5(C+N)) e 347 (0,08C+Nb≥10C), são dadas a seguir. Nota-se que um aço
AISI 347 apresenta uma resistência sensivelmente superior a de um aço AISI 321, como
mostrado na Figura 2.31 [39].
Figura 2.31: Comparativo entre curvas TTP para os aços AISI 304, 304L, 321 e 347.
[39]
Observa-se que, devido aos curtos intervalos de tempo necessários à precipitação do
carboneto na faixa de temperaturas entre ∼650 e 870ºC, um aço 304 pode se sensitizar
na ZTA (ou sofrer o ″weld decay″) por decorrência da aplicação de um simples ciclo
térmico de soldagem, ao passo que um 304L (com baixo carbono) não. Por sua vez, os
aços AISI 321 e 347 são ditos ″estabilizados″. Esses aços possuem em sua formulação
elementos, Ti e Nb respectivamente, que possuem maior afinidade com o carbono do
que o cromo, formando-se os carbonetos TiC ou NbC ao invés do (Cr-Fe)23C6, o que
possibilita o emprego contínuo desses materiais à temperaturas próximas a 500°C (o

51 AISI 347, principalmente), sem que hajam riscos de sensitização (as especificações
dos teores de Ti e Nb devem obedecer às relações vistas acima). Entretanto, a soldagem
pode deixar o MB, na imediata adjacência à linha de fusão, vulnerável à corrosão
devido a solubilização dos elementos estabilizantes associada a uma taxa de
resfriamento que impeça a reformação do TiC/NbC (″knife-line attack″). Neste caso, a
precipitação do (Cr-Fe)23C6, nestas posições, pode se dar em serviço por exemplo. [32]
Não se conhece um único mecanismo que possa responder por todas as ocorrências de
corrosão sob tensão. Não considerando as ligas que teriam (qualquer que fosse a razão)
definidas as trajetórias para a propagação (tal como uma microestrutura sensitizada) e
aquelas fragilizadas pelo hidrogênio, os mecanismos pelos quais uma trinca poderia
propagar-se num ambiente quimicamente agressivo, são [40]:
- propagação por clivagem em filmes frágeis (″film induced cleavage″). Uma trinca se
propagaria por repetidas etapas de formação e ruptura de um filme de natureza frágil
que se desenvolve na extremidade da trinca. A extensão da propagação em cada
etapa poderia ou não permanecer confinada ao filme;
- propagação por mobilidade superficial (″surface mobility″). A propagação se daria
por captura de lacunas ou vazios na extremidade da trinca, fazendo-se necessário a
auto-difusão superficial, como apresentado na Figura 2.32. Reporta-se que a
contaminação da superfície pode gerar vazios e acelerar ou retardar a mobilidade
superficial, e que os contaminantes que aceleram essa mobilidade são justamente
aqueles conhecidos por induzir a corrosão sob tensão;
Figura 2.32: Representação esquemática do mecanismo de propagação por ″mobilidade
superficial″.[40].
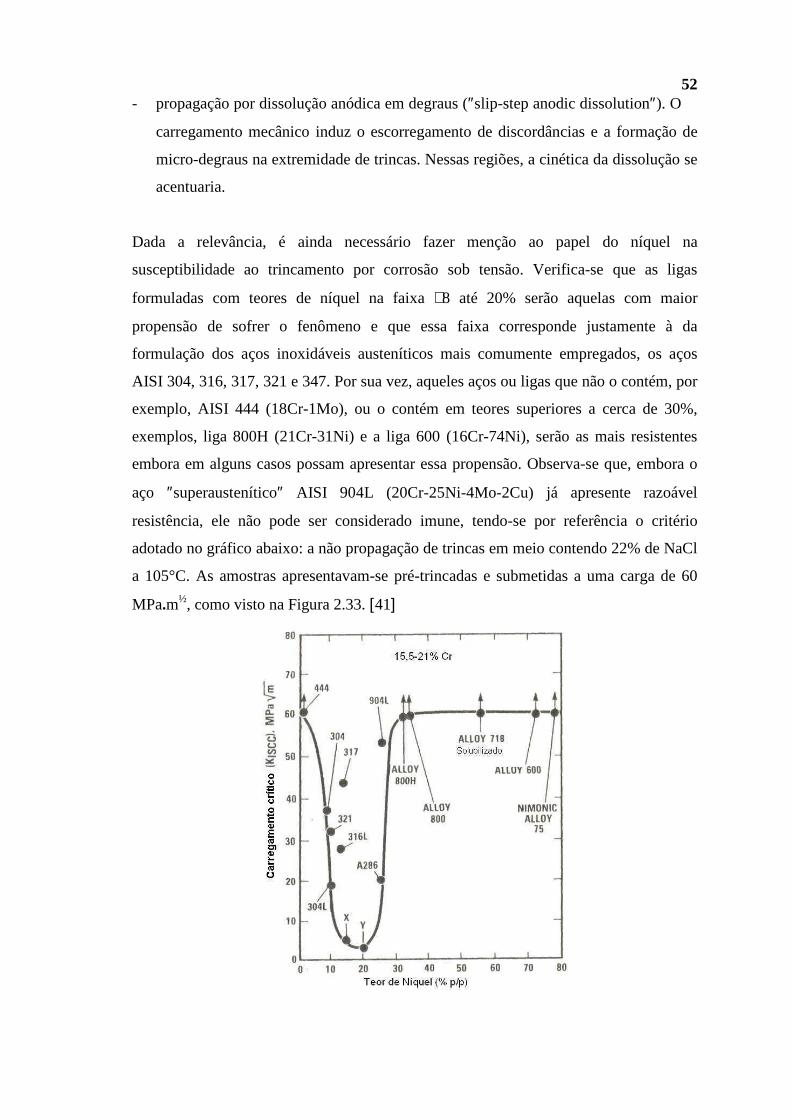
52 - propagação por dissolução anódica em degraus (″slip-step anodic dissolution″). O
carregamento mecânico induz o escorregamento de discordâncias e a formação de
micro-degraus na extremidade de trincas. Nessas regiões, a cinética da dissolução se
acentuaria.
Dada a relevância, é ainda necessário fazer menção ao papel do níquel na
susceptibilidade ao trincamento por corrosão sob tensão. Verifica-se que as ligas
formuladas com teores de níquel na faixa ∼8 até 20% serão aquelas com maior
propensão de sofrer o fenômeno e que essa faixa corresponde justamente à da
formulação dos aços inoxidáveis austeníticos mais comumente empregados, os aços
AISI 304, 316, 317, 321 e 347. Por sua vez, aqueles aços ou ligas que não o contém, por
exemplo, AISI 444 (18Cr-1Mo), ou o contém em teores superiores a cerca de 30%,
exemplos, liga 800H (21Cr-31Ni) e a liga 600 (16Cr-74Ni), serão as mais resistentes
embora em alguns casos possam apresentar essa propensão. Observa-se que, embora o
aço ″superaustenítico″ AISI 904L (20Cr-25Ni-4Mo-2Cu) já apresente razoável
resistência, ele não pode ser considerado imune, tendo-se por referência o critério
adotado no gráfico abaixo: a não propagação de trincas em meio contendo 22% de NaCl
a 105°C. As amostras apresentavam-se pré-trincadas e submetidas a uma carga de 60
MPa.m½, como visto na Figura 2.33. [41]

53 Figura 2.33. Gráfico ilustrativo do papel do níquel na propensão que várias ligas pré-
trincadas apresentam de ter trincas propagadas por corrosão sob tensão
em solução contendo 22% NaCl a 105°C. Tais resultados sugerem como
imunes a corrosão sob tensão as ligas cujas trincas não propagam com
carregamentos iguais ou superiores a 60MPa.m½. As ligas X e Y se
referem à ligas de procedência alemã resistentes ao calor. [41]

54 CAPÍTULO 3
3.1 O serviço em baixa e em alta temperatura
Subjacente a essas condições ″extremas″ de serviço, uma envolvendo baixa temperatura
e outra temperaturas elevadas, encontram-se mecanismos fundamentais associados com
a redução da tenacidade com a redução da temperatura e com a deformação induzida
por fluência a alta temperatura, ambas fazendo uso de argumentos envolvendo defeitos
de rede que são encontrados em profusão na estrutura cristalina dos aços.
Por fluência, entende-se a deformação sofrida pela peça ou componente de um dado
equipamento com o tempo, o que se verifica sempre que, fixada a metalurgia, se tenha
uma solicitação conjunta temperatura+carregamento mecânico em nível adequado. Pode
culminar com perdas da funcionalidade por distorção ou mesmo com a ruptura da peça
ou componente. É o que tende a ocorrer, por exemplo, nas colunas dos fornos de
reforma ou na tubulação por onde flui vapor superaquecido de alta pressão,
∼61kgf/cm2ab a 440ºC (essas são as características do ″vapor de alta″ produzido na
Regap).
Por outro lado, observa-se que um comportamento frágil do aço não é induzido apenas
por baixas temperaturas; altas temperaturas promotoras da precipitação de fases
intermetálicas fragilizantes (por exemplo, a fase ″sigma″ nos aços inoxidáveis), podem
também desencadear um comportamento frágil à temperatura ambiente. Do mesmo
modo, assim tendem a se comportar as peças espessas contendo descontinuidades
lineares pelo fato de que nesses casos, usualmente, associa-se um estado de tensões
(plano de deformação) inibidor da ativação dos mecanismos de deformação plástica;
assim também se comportam peças de aço hidrogenadas pelo contato com o H2S em
meio aquoso, e ainda os aços submetidos a carregamentos realizados com altas taxas de
deformação.
Por outro lado, as condições reinantes numa grande variedade de situações encontradas
em refinarias, envolvem temperaturas nas quais, além da deformação por fluência,
pode-se ter alterações microestruturais que degradam as propriedades mecânicas. É o
caso da ″grafitização″ do aço carbono. Adicionalmente, por conseqüência da interação
com meios reativos, as propriedades ″anti-corrosivas″ do aço podem deteriorar-se.

55 Serve como exemplo a carbonetação do aço inoxidável decorrente do contato com
misturas gasosas compostas por CO e CO2 a altas temperaturas (∼700ºC), em refinarias
um ambiente originado da regeneração do catalisador em UFCC’s, ou ainda ter-se a
descarbonetação interna e a fissuração do aço ferrítico provocado por misturas H2S+H2
a temperaturas intermediárias (350ºC), meio (H2S) oriundo das reações catalisadas de
dessulfurização do hidrocarboneto em unidades HDT.
3.1.1 Baixas temperaturas. Aspectos microscópicos e microestruturais
Em refinarias, o armazenamento sob pressão do GLP à temperatura ambiente é feita em
vasos de pressão esféricos ou ″esferas″. Esse armazenamento envolve condições sob as
quais a possibilidade de atingir-se baixas temperaturas nas paredes deve ser observada
em projeto. Tal fato poderia se dar por descompressão acidental do gás liqüefeito
(vazamento em ligações flangeadas, por exemplo), caso em que a temperatura do
hidrocarboneto deve cair até que se atinja a temperatura de ebulição do líquido
remanescente na pressão na qual a estanqueidade seja retomada. Essa temperatura pode
ser muito baixa; por exemplo, à pressão atmosférica, o C3H8 entra em ebulição a –
42,3ºC e o CH4, o principal componente do gás natural, a –160ºC. [42, 43]
Os aços ferríticos ou os metais e ligas estruturadas por redes CCC caracterizam-se por
oferecer uma maior resistência ao ″escorregamento de discordâncias″ do que os metais e
ligas estruturados por redes CFC. Serve como exemplo dessas últimas os aços
inoxidáveis austeníticos da série 300.
Discordâncias são defeitos associados a existência de planos incompletos de átomos, os
quais em sua terminação, dão origem a uma ″linha″ de átomos. Além desse ponto, o
vazio deixado pelas linhas ausentes são preenchidos pelas filas vizinhas, que se
deformam para se aproximarem. São as discordâncias em ″aresta″ (cuja geometria
difere das discordâncias em ″hélice″). Tais defeitos se revelam como os principais
responsáveis pela plasticidade apresentadas pelos metais e ligas à temperatura ambiente,
a baixas temperaturas e, sob certas condições, a altas temperaturas. A uma dada
temperatura, quando o aço é solicitado mecanicamente por um carregamento em um
nível que acarrete sua deformação plástica, ela irá decorrer do escorregamento de uma
enorme quantidade desses defeitos sobre planos cristalográficos específicos,

56 considerando-se que cada um deles é capaz de ocasionar o cisalhamento da micro-
região adjacente. No aço carbono recozido existem cerca de 108/cm2 dessas
imperfeições; este mesmo aço trabalhado a frio passa a conter ~1011/cm2. Entretanto,
deve-se observar que, neste caso, a interação mútua entre discordâncias acaba por inibir
o próprio escorregamento, o que termina por tornar o aço menos deformável
plasticamente. Tornam-se, desta forma, menos dúcteis e menos macios. [44, 45]
Mostra-se a seguir, na Figura 3.1, os aspectos micro-fractográficos associados a uma
superfície de fratura produzida por mecanismo dúctil. A fratura se dá pela formação e
coalescência de micro-cavidades hemisféricas ou parabólicas (″dimples″), ambos os
fenômenos dependentes da ativação dos mecanismos de deformação plástica. A
característica macroscópica correspondente é a ″fibrosa″, pouco brilhante ou opaca.
Figura 3.1: Característica apresentada por uma superfície metálica que fraturou de
forma dúctil. (MEV-1500×). [20]
A resistência imposta pelo reticulado cristalino ao escorregamento das discordâncias,
dá-se sob a forma de um "atrito interno" (″lattice friction″), gerado por forças oriundas
de um desequilíbrio que se verifica existir entre os átomos adjacentes a ela. Fixada a
temperatura, a tensão τ necessária para vencê-lo, a ser produzida por carregamento
externo, reduz-se exponencialmente com a ″largura″ w da discordância; é a tensão de
Peierls-Nabarro, τ ∝ e-w . Assim, quanto maior for o número de átomos envolvidos (ou
seja, maior for o w), menor deverá ser esse atrito, o que seria devido a um cancelamento
mais eficaz dessas forças produzidas por um maior número de pares de átomos

57 localizados simetricamente em torno da discordância. Deduz-se daí, que as
discordâncias existentes nos metais CFC, estrutura cujo arranjo de átomos é mais
compacto do que o apresentado por arranjos CCC, devam possuir maiores w′s,
justificando-se, em parte (um maior número de planos de escorregamento também deve
contribuir), a tenacidade e a ductilidade superiores dos aços austeníticos (CFC)
relativamente aos aços ferríticos (CCC). [45]
Adicionalmente, reduções da temperatura acarretam o aumento do atrito a ser vencido
por discordâncias de pequeno w, ou seja, aquelas existentes em metais CCC. Tal
fenômeno parece indicar que a redução da amplitude vibracional dos átomos com a
temperatura acentua esse efeito, particularmente nessas redes. Assim, tais reduções
terminam por proporcionar o aumento da tensão de Peierls-Nabarro, fato que, por sua
vez, irá se refletir no aumento do limite de escoamento σy do aço (tensão a partir da qual
inicia-se a deformação plástica ou o escorregamento de discordâncias). Por outro lado, a
transição de comportamento dúctil-frágil em um metal CCC (os metais CFC não
apresentam esse fenômeno, mantendo-se dúcteis mesmo a muito baixas temperaturas), é
atribuída à formação de micro-trincas em antecedência a fratura. Essa pré-condição é
também encontrada na teoria de Griffith para materais cujo comportamento é puramente
elástico. As micro-trincas seriam precedidas e ocasionadas por deformação plástica
envolvendo o escorregamento, o acúmulo (″pile-up″) e a ″coalescência″ de
discordâncias em barreiras tais como são os contornos de grão. Sendo a nucleação de
micro-trincas um fenômeno que deve ocorrer toda vez que a tensão de escoamento
superar a tensão oriunda da interação entre discordâncias próximas, parece natural que
exista algo como uma temperatura de transição, e que ela se dê a baixas temperaturas
(pois nestas condições verifica-se o aumento do σy). Além do mais, o crescimento
dessas trincas só se dará se a tensão necessária para que esse crescimento se dê for
inferior a tensão de escoamento. Então, pode-se concluir, que quanto maior for o σy
(maior for a redução de temperatura), mais provável será ter-se um comportamento
frágil. [45]
O conjunto de figuras dado a seguir busca evidenciar: o acúmulo de discordâncias no
contorno de grão, Figura 3.2a, a formação de micro-trincas que antecedem um
comportamento frágil, Figura 3.2b, e, tal como na Figura 3.1, os aspectos micro-
fractográficos associados a uma fratura frágil, Figura 3.2c.

58
(a) (b)
(c)
Figuras 3.2: (a) Evidência do acúmulo de discordâncias em interface (barreira)
produzida por contorno de grão em uma fina lâmina de aço inoxidável.
(17.500x, MET) [44], (b) Micro-trincas produzidas por carregamento em
tração em uma lâmina de ferro a −140ºC. (250×, MEV) [44] e, (c)
Facetas de clivagem tipicamente encontradas na superfície dos metais
fraturados fragilmente. Tais superfícies tendem a apresentar um aspecto
brilhante ou ″cristalino″ a vista desarmada. (1560×-MEV). [20]
Existem evidências de que a etapa de propagação das micro-fissuras constituí-se na
controladora do fenômeno, uma vez que os contornos de grão (desde que não se
encontrem fragilizados), formam barreiras muito efetivas, dificultando essa propagação.
Assim, o tamanho de grão (TG) cumpre importante papel na tenacidade, e o seu refino,
por aumentar a extensão dessas barreiras, contribui de forma efetiva na melhoria dessa
propriedade. Petch foi capaz de encontrar a relação que há entre o diâmetro do grão, d, e
a tensão σf necessária para a propagação, σf ∝ d−1/2. [44]
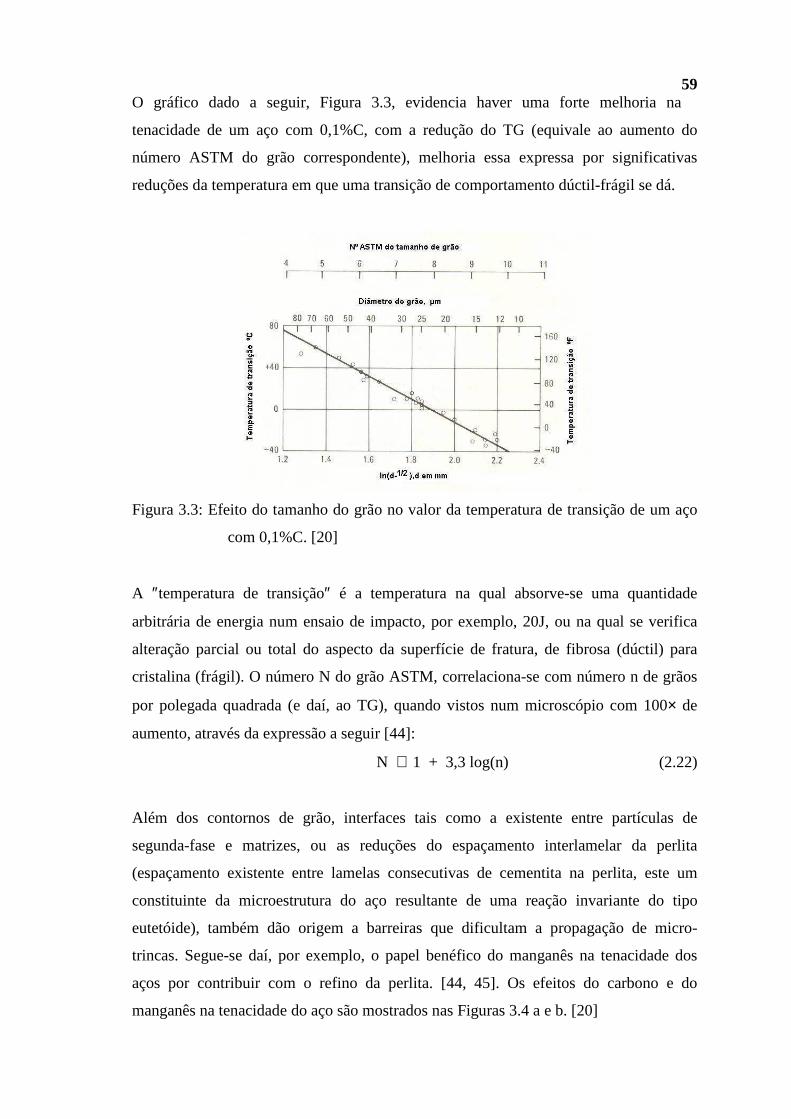
59 O gráfico dado a seguir, Figura 3.3, evidencia haver uma forte melhoria na
tenacidade de um aço com 0,1%C, com a redução do TG (equivale ao aumento do
número ASTM do grão correspondente), melhoria essa expressa por significativas
reduções da temperatura em que uma transição de comportamento dúctil-frágil se dá.
Figura 3.3: Efeito do tamanho do grão no valor da temperatura de transição de um aço
com 0,1%C. [20]
A ″temperatura de transição″ é a temperatura na qual absorve-se uma quantidade
arbitrária de energia num ensaio de impacto, por exemplo, 20J, ou na qual se verifica
alteração parcial ou total do aspecto da superfície de fratura, de fibrosa (dúctil) para
cristalina (frágil). O número N do grão ASTM, correlaciona-se com número n de grãos
por polegada quadrada (e daí, ao TG), quando vistos num microscópio com 100× de
aumento, através da expressão a seguir [44]:
N ≅ 1 + 3,3 log(n) (2.22)
Além dos contornos de grão, interfaces tais como a existente entre partículas de
segunda-fase e matrizes, ou as reduções do espaçamento interlamelar da perlita
(espaçamento existente entre lamelas consecutivas de cementita na perlita, este um
constituinte da microestrutura do aço resultante de uma reação invariante do tipo
eutetóide), também dão origem a barreiras que dificultam a propagação de micro-
trincas. Segue-se daí, por exemplo, o papel benéfico do manganês na tenacidade dos
aços por contribuir com o refino da perlita. [44, 45]. Os efeitos do carbono e do
manganês na tenacidade do aço são mostrados nas Figuras 3.4 a e b. [20]
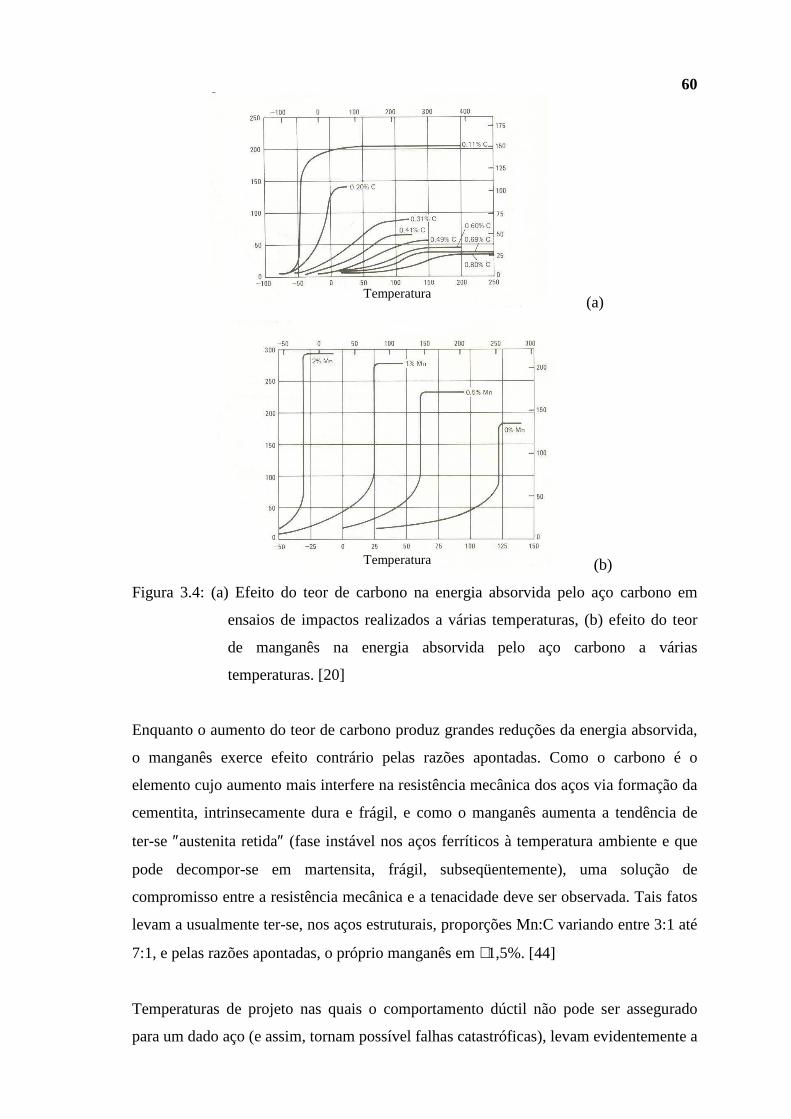
60
(a)
(b)
Figura 3.4: (a) Efeito do teor de carbono na energia absorvida pelo aço carbono em
ensaios de impactos realizados a várias temperaturas, (b) efeito do teor
de manganês na energia absorvida pelo aço carbono a várias
temperaturas. [20]
Enquanto o aumento do teor de carbono produz grandes reduções da energia absorvida,
o manganês exerce efeito contrário pelas razões apontadas. Como o carbono é o
elemento cujo aumento mais interfere na resistência mecânica dos aços via formação da
cementita, intrinsecamente dura e frágil, e como o manganês aumenta a tendência de
ter-se ″austenita retida″ (fase instável nos aços ferríticos à temperatura ambiente e que
pode decompor-se em martensita, frágil, subseqüentemente), uma solução de
compromisso entre a resistência mecânica e a tenacidade deve ser observada. Tais fatos
levam a usualmente ter-se, nos aços estruturais, proporções Mn:C variando entre 3:1 até
7:1, e pelas razões apontadas, o próprio manganês em ∼1,5%. [44]
Temperaturas de projeto nas quais o comportamento dúctil não pode ser assegurado
para um dado aço (e assim, tornam possível falhas catastróficas), levam evidentemente a
Temperatura
Temperatura

61 inadequação desse material. Para os aços C-Mn, essa condição pode ser encontrada à
temperaturas inferiores a - 45ºC. São os serviços criogênicos. Neste caso, os aços
ligados ao níquel ou mesmo aqueles com matrizes austeníticas (aço inoxidável da série
300), passam a ser necessários [43]. Um exemplo das conseqüências de uma falha
catastrófica pode ser vista na foto da Figura 3.5.
Figura 3.5: Conseqüências da liberação abrupta de energia devido ao rompimento frágil
do fundo de um tanque durante execução de teste pneumático. (Refap,
RS).
Neste caso, a despressurização repentina do ar armazenado à pressão p em um tanque de
volume V e peso mg durante execução de um teste pneumático realizado à temperatura
ambiente, dá-se devido a ruptura de uma grande extensão de seu fundo causada por
sobrecarga na pressão. Observar na foto, a deformação sofrida pelo tanque em sua
metade superior, o que não ocorre na metade inferior, possivelmente pela maior rigidez
conferida pelo sistema de fixação do fundo em base de alvenaria, o que teria induzido
um comportamento frágil daquela região. A grosso modo, pode-se afirmar que a energia
liberada no processo (pV) se conserva transformando-se em potencial (mgh), o que
acarreta a ejeção do tanque a uma altura da ordem de:
h =p.V
m.g (2.23)

62 Não obstante, não tratar-se de um aço e, sim, de uma liga a base de níquel (Inconel
625), dada como ″não endurecível por precipitação″ por tratamentos térmicos, o
exemplo mostrado a seguir, na Figura 3.6, serve para ilustrar o rompimento frágil de
uma liga que originalmente era dúctil o suficiente para ser selecionada para a fabricação
de foles de juntas de expansão empregadas em UFCC’s. Falha ocorrida durante parada
(resfriamento) da unidade e decorrente da perda da capacidade da liga de suportar as
solicitações mecânicas existentes nesses períodos. Verificou-se ter havido uma muito
importante redução da ductilidade e aumento da dureza ao expô-la de forma contínua ao
serviço que envolvia temperaturas da ordem de 700ºC, o que levou a ter-se uma intensa
precipitação da fase γ″ (Ni3Nb). [46]
Figura 3.6: Ruptura frágil de uma das corrugações de um fole (∅42″-2 mm de parede)
de uma junta de expansão de uma UFCC fabricado em Inconel 625
(ASTM B-443), Ni-20Cr-8Mo-4Nb. (Regap, MG).

63
3.1.2 Altas temperaturas
3.1.2.1 Fluência
Como foi indicado, sempre que se tiver uma solicitação conjunta
temperatura+carregamento mecânico, ter-se-á em maior ou em menor nível, a depender
da intensidade do carregamento, a deformação do aço que tende a se intensificar com o
tempo.
Para os aços C e C-Mn (para referência, considerá-los como material construtivo dos
tubos de especificação ASTM A-53B e A-106B, respectivamente), a fluência deve,
necessariamente, ser levada em conta no projeto à temperaturas superiores a cerca de
420°C, independentemente da natureza do meio. Para esses aços, essa é a temperatura a
partir da qual as tensões admissíveis para fluência para uma vida de projeto de 100.000h
(curva 6) Figura 3.6: Ruptura frágil de uma das corrugações de um fole (∅42″-2 mm de
parede) de uma junta de expansão de uma UFCC fabricado em Inconel 625 (ASTM B-
443), Ni-20Cr-8Mo-4Nb. (Regap, MG).
tornam-se inferiores à tensão admissível elástica (curva 5), e assim devem ser as
consideradas, Figura 3.7. [47]

64
Figura 3.7: Conjunto de curvas empregadas no dimensionamento da espessura de parede
dos tubos fabricados nos aços A-106B e A-53B para aplicação em temperatura
elevada. No gráfico, a numeração encontrada nas várias curvas indicam: 1.
limite de resistência mín., 2. variação do limite de resistência com a
temperatura, 3. limite de escoamento mín., 4. variação do limite de
escoamento com a temperatura, 5. tensão admissível para o campo elástico, 6.
tensão admissível para fluência para uma vida de 20, 40, 60 e 100.000h, 7.
temperatura limite de projeto para os materiais referenciados, ∼540°C, 8.
tensão de ruptura mínimo, 9. tensão de ruptura médio, 10. projeto no campo
elástico para tensões superiores à indicada, 110MPa. [47]
Enquanto o lado esquerdo do gráfico permite a obtenção direta da tensão admissível Sa
para uma vida de projeto L fixada a temperatura (de projeto) T, o seu lado direito
correlaciona o carregamento mecânico σ com o ″parâmetro P″ de Larsen-Miller. Essa é
uma técnica paramétrica que presume que a razão entre a energia de ativação ∆H para a
fluência (barreira de energia a ser superada por um átomo se deslocar de uma posição
para outra com menor energia) e a constante dos gases R, ∆H/R, é constante e
independe da temperatura e da tensão aplicadas. Então, o tempo para que uma ruptura
por fluência se dê variará com a temperatura de tal forma que P, P( ≡ ∆H/R), expresso

65 por T (20+logL), permaneça invariável. Consegue-se assim, reunir em uma única reta
uma grande quantidade de dados obtidos de ensaios acelerados de fluência, permitindo
extrapolar, a partir desses resultados, qual deverá ser a vida L do componente em
condições industriais, fixada as características metalúrgicas. [48]
Os mecanismos que produzem a deformação por fluência podem envolver tanto a a
difusão de espécies atômicas (lacunas e átomos), através do grão (mecanismo Nabarro-
Herring), ou pelo seu contorno (mecanismo Coble), como a ″escalagem″ de
discordâncias (fenômeno também dependente da movimentação de átomos ou lacunas).
A mobilidade dessas espécies é descrita pelo coeficiente de difusão D no meio (isto é,
na estrutura cristalina do aço), o qual correlaciona-se com a temperatura T por uma
relação do tipo Arrhenius, D ∝ exp(−1/T). Por sua vez, a taxa de deformação dε/dt,
quando se tem associadas baixas tensões σ e altas temperaturas T guarda a
proporcionalidade, dε/dt ∝ (σ/Td2), onde d é diâmetro do grão. Dessa forma, fixada a
temperatura e o carregamento, o aumento do grão, ou como é equivalente, a redução de
seu contorno, traria como resultado imediato um efeito retardador dessas deformações e
uma maior resistência à fluência do aço com uma granulação mais grosseira. Assim, se
verifica que o tamanho de grão em fluência exerce efeito oposto ao exercido na
tenacidade. [45, 48]
O ″mapa de deformação″ de Ashby apresentado na Figura 3.8, para um aço alta liga Fe-
20Cr-30Ni com tamanho de grão médio igual a 200µm, permite identificar o
mecanismo de deformação predominante nos vários domínios. Além do mecanismo, o
mapa fornece as taxas de deformação em cada caso e evidencia que a fluência por
difusão coincide com as condições tensão–temperatura (~1 a ~10MN/m2, T≈0,7Tfusão)
encontradas em aplicações na indústria envolvendo esse material particularmente.
Entretanto, essa é uma característica encontrada na quase totalidade das aplicações na
indústria. Devido às altas taxas de deformação envolvidas, quando o projeto envolve
componentes que em serviço estarão sujeitos à fluência, é usual procurar-se um
dimensionamento que minimize tensões, evitando-se tornar operativo mecanismos de
deformação que envolvam o escorregamento de discordâncias. [49, 50]

66
Figura 3.8: Mapa de deformação de Ashby para um aço Fe-20Cr-30Ni. Notar a
predominância dos mecanismos de deformação de Coble sob condições
que envolvem a aplicação industrial da liga. [49]
Até algum tempo atrás, os tubos ou as ″colunas″ (obtidos por fundição centrífuga)
empregados em fornos de reforma a vapor geradores de hidrogênio eram, muito
freqüentemente, especificados em aços A608GrHK-40 (25Cr-20Ni-0,4C). Tais aços
possuem matriz austenítica compostas por grãos de morfologia colunar orientados
perpendicularmente à parede do tubo, nos quais distribuem-se carbonetos eutéticos
interdendríticos ditos primários. Figura 3.9a. Colocados em serviço, com o tempo e à
temperaturas superiores a 650°C, novos carbonetos altamente dispersos, ditos
secundários, passam a precipitar e compor, juntamente com os primários, a
microestrutura do aço, como mostrado na Figura 3.9b. Uma vez que os ″vazios″
promovidos por fluência tendem a nuclear nas interfaces matriz-carbonetos primários
coalescidos (isto é, uma rede contínua de carbonetos), a resistência que o aço oferece à
fluência é diretamente associada à resistência que os carbonetos oferecem ao
coalescimento. Neste caso, a microestrutura passa a mostrar intensa presença de
carbonetos secundários dispersos (nesta condição é dito envelhecido e a
ductilidade/soldabilidade tendem a ″zero″); apresenta também uma rede de carbonetos
primários e vazios de fluência (pontos escuros) nucleados na interface, os quais já
apresentam uma tendência ao alinhamento. [51]
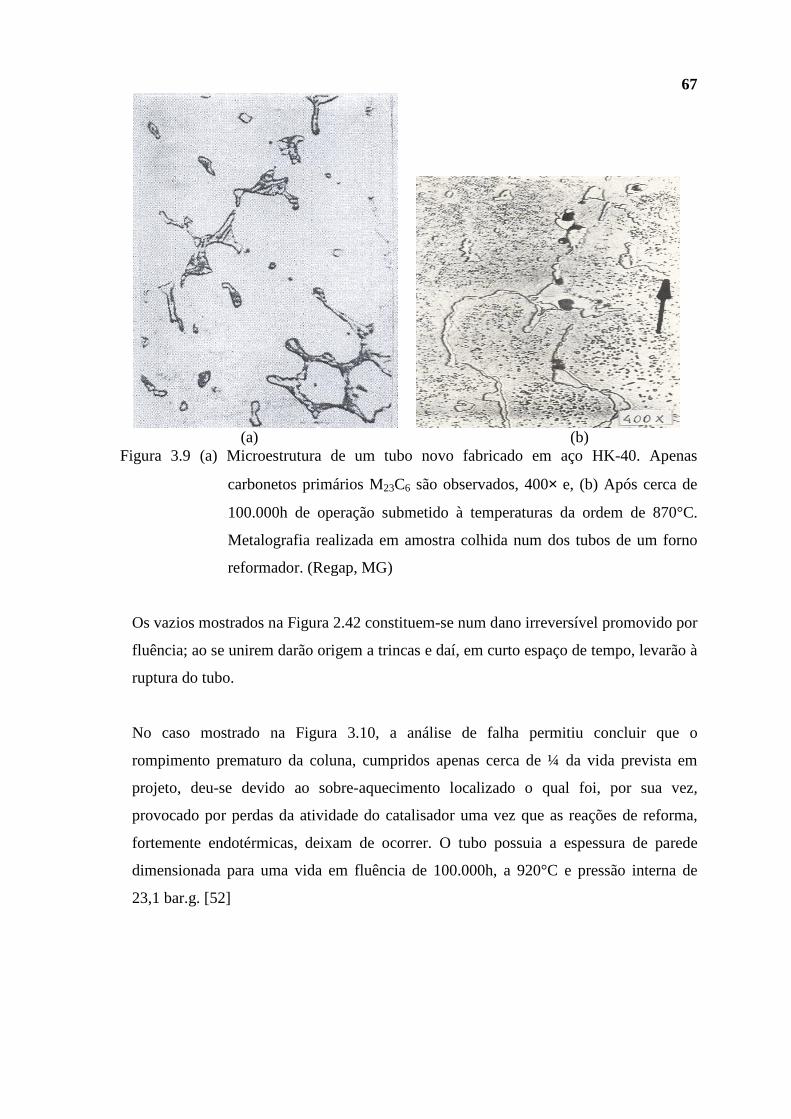
67
(a) (b)
Figura 3.9 (a) Microestrutura de um tubo novo fabricado em aço HK-40. Apenas
carbonetos primários M23C6 são observados, 400× e, (b) Após cerca de
100.000h de operação submetido à temperaturas da ordem de 870°C.
Metalografia realizada em amostra colhida num dos tubos de um forno
reformador. (Regap, MG)
Os vazios mostrados na Figura 2.42 constituem-se num dano irreversível promovido por
fluência; ao se unirem darão origem a trincas e daí, em curto espaço de tempo, levarão à
ruptura do tubo.
No caso mostrado na Figura 3.10, a análise de falha permitiu concluir que o
rompimento prematuro da coluna, cumpridos apenas cerca de ¼ da vida prevista em
projeto, deu-se devido ao sobre-aquecimento localizado o qual foi, por sua vez,
provocado por perdas da atividade do catalisador uma vez que as reações de reforma,
fortemente endotérmicas, deixam de ocorrer. O tubo possuia a espessura de parede
dimensionada para uma vida em fluência de 100.000h, a 920°C e pressão interna de
23,1 bar.g. [52]

68
Figura 3.10: Ruptura axial por fluência em tubo (A-608GrHK-40/∅5″) de um forno de
reforma. (Regap, MG)
Na maior parte das situações que envolvem temperaturas entre ∼450 e ∼700ºC, os aços
ligados ao Cr e ao Mo são as principais opções. As razões da seleção desses aços são: a)
ao contrário do aço carbono, são imunes à grafitização pois os carbonetos de molibdênio
e de cromo são mais estáveis do que o de ferro, b) possuem superior resistência a
oxidação devido à proteção dada pela presença de óxidos e espinélios superficiais a base
de Fe e Cr e, c) oferecem adequada resistência à deformação por fluência. Tal
característica vem como resultado conjunto do efeito endurecedor por solução sólida
conferido tanto pelo molibdênio como pelo cromo, da precipitação dos carbonetos dos
mesmos elementos que se distribuem com grande dispersão na matriz ferrítica,
constituindo-se em obstáculos efetivos para o escorregamento de discordâncias,
podendo também precipitar nos contornos, neste caso, dificultando também o
deslizamento de grãos contíguos. [48, 53]
Assim, enquanto que para a fluência (apenas fluência) um aço C-Mn tem sua aplicação
limitada a 540ºC, um aço liga do tipo 21/4Cr-1Mo pode ser especificado para o serviço
contínuo até 650°C e um 7Cr-1/2Mo até 705ºC [47]. Os efeitos do Mo e do Cr no
aumento da resistência à fluência (tensão para a ruptura em até 1000h) são mostrados a
seguir na Figura 3.11.
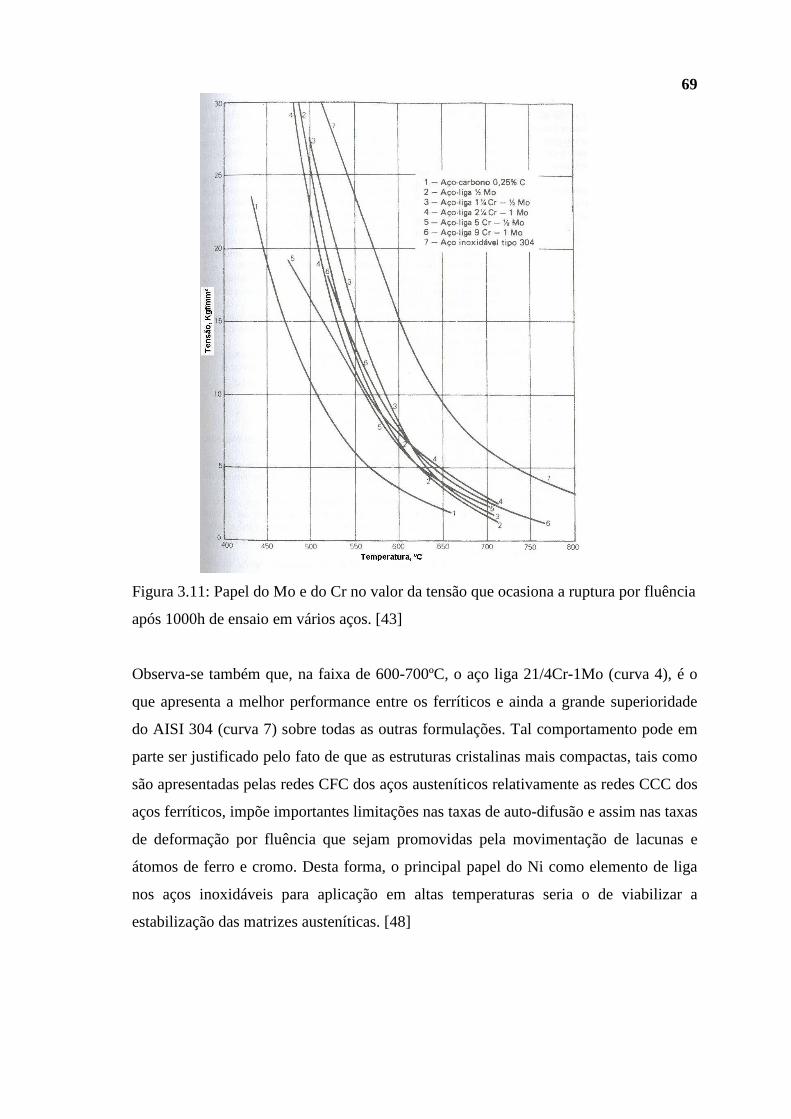
69
Figura 3.11: Papel do Mo e do Cr no valor da tensão que ocasiona a ruptura por fluência
após 1000h de ensaio em vários aços. [43]
Observa-se também que, na faixa de 600-700ºC, o aço liga 21/4Cr-1Mo (curva 4), é o
que apresenta a melhor performance entre os ferríticos e ainda a grande superioridade
do AISI 304 (curva 7) sobre todas as outras formulações. Tal comportamento pode em
parte ser justificado pelo fato de que as estruturas cristalinas mais compactas, tais como
são apresentadas pelas redes CFC dos aços austeníticos relativamente as redes CCC dos
aços ferríticos, impõe importantes limitações nas taxas de auto-difusão e assim nas taxas
de deformação por fluência que sejam promovidas pela movimentação de lacunas e
átomos de ferro e cromo. Desta forma, o principal papel do Ni como elemento de liga
nos aços inoxidáveis para aplicação em altas temperaturas seria o de viabilizar a
estabilização das matrizes austeníticas. [48]

70 3.1.2.2 Fadiga-térmica e o colapso incremental
Os cascos dos reatores das unidades de coqueamento retardado, equipamentos que
invariavelmente se apresentam sempre em número par de forma a viabilizar uma
operação sincronizada de carregamento e descarregamento, são compostos por aneis de
diferentes espessuras soldados entre si em aço baixa liga Cr-Mo. Esses anéis são
revestidos internamente com aço inoxidável, freqüentemente um aço inoxidável (12Cr),
dada a temperatura e a significativa presença do H2S na carga (resíduo efluente da torre
fracionadora da planta), considerando a resistência que esse material oferece à corrosão
por sulfetação.
Esses equipamentos são submetidos em serviço, além da alternância da pressão interna,
a um severo regime de tensões térmicas também cíclicas. É, assim, ciclicamente
solicitado por uma combinação de carregamentos P+Q, onde P desígna um
carregamento primário e Q um carregamento secundário.
Um carregamento primário diz respeito ao campo de tensões que se produz nas paredes
do equipamento decorrentes do equilíbrio que se estabelece em resposta às forças
externas exercidas sobre a estrutura (pressão interna ou peso próprio, por exemplo). O
secundário é associado à tensões térmicas produzidas por gradientes de temperaturas, ou
pela união de metais igualmentes aquecidos mas possuidores de diferentes coeficientes
de dilatação, ou ainda pela existência de restrições geométricas em pontos de transição
de forma a impedir ou dificultar a deformação do metal adjacente (transição casco
cilíndrico-tampo toriesférico dos vasos de pressão submetidos à pressão interna, por
exemplo). [54]
Os carregamentos secundários (tal como as tensões residuais em soldas), caracterizam-
se por se auto-limitarem ao nível do limite de escoamento σy do aço; é o fenômeno de
relaxação espontâneo de tensões (denominado ″shakedown″). Esse comportamento não
é observado nos carregamentos primários cujo campo de tensão gerado na estrutura não
se limita com a deformação da mesma. Dessa forma, os carregamentos primários
podem, potencialmente, ocasionar o escoamento generalizado e por fim a ruptura das
paredes do equipamento se não for aliviado ou reduzido. É o modo de falha por "ruptura
dúctil". Tal modo de falha é prevenido ao limitar-se a tensão admissível Sa para um
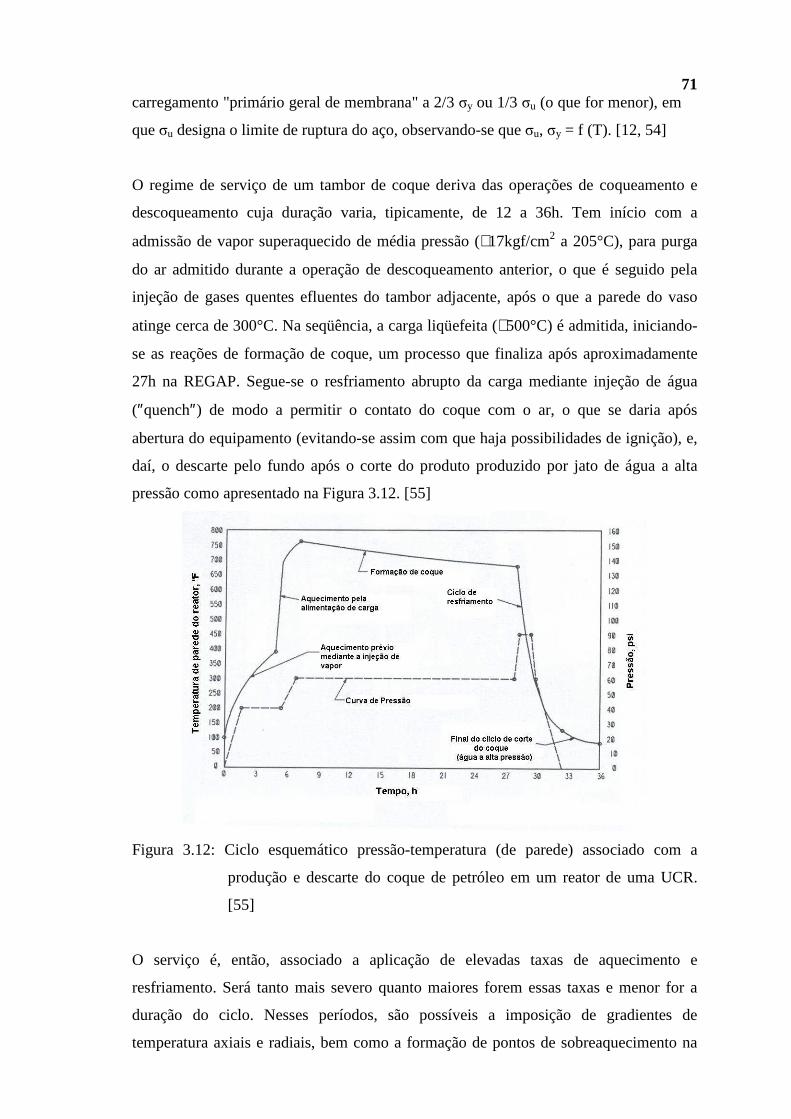
71 carregamento "primário geral de membrana" a 2/3 σy ou 1/3 σu (o que for menor), em
que σu designa o limite de ruptura do aço, observando-se que σu, σy = f (T). [12, 54]
O regime de serviço de um tambor de coque deriva das operações de coqueamento e
descoqueamento cuja duração varia, tipicamente, de 12 a 36h. Tem início com a
admissão de vapor superaquecido de média pressão (∼17kgf/cm2 a 205°C), para purga
do ar admitido durante a operação de descoqueamento anterior, o que é seguido pela
injeção de gases quentes efluentes do tambor adjacente, após o que a parede do vaso
atinge cerca de 300°C. Na seqüência, a carga liqüefeita (∼500°C) é admitida, iniciando-
se as reações de formação de coque, um processo que finaliza após aproximadamente
27h na REGAP. Segue-se o resfriamento abrupto da carga mediante injeção de água
(″quench″) de modo a permitir o contato do coque com o ar, o que se daria após
abertura do equipamento (evitando-se assim com que haja possibilidades de ignição), e,
daí, o descarte pelo fundo após o corte do produto produzido por jato de água a alta
pressão como apresentado na Figura 3.12. [55]
Figura 3.12: Ciclo esquemático pressão-temperatura (de parede) associado com a
produção e descarte do coque de petróleo em um reator de uma UCR.
[55]
O serviço é, então, associado a aplicação de elevadas taxas de aquecimento e
resfriamento. Será tanto mais severo quanto maiores forem essas taxas e menor for a
duração do ciclo. Nesses períodos, são possíveis a imposição de gradientes de
temperatura axiais e radiais, bem como a formação de pontos de sobreaquecimento na

72 superfície metálica devido a existência de irregularidades aleatórias na distribuição
do coque, o qual adere à parede dos reatores [56]. A parede é, assim, sistematicamente
submetida a uma complexa, intensa e não necessariamente semelhante (ciclo a ciclo),
distribuição de tensões térmicas.
Para um carregamento do tipo P+Q cíclico, recomenda-se o atendimento da
desigualdade P+Q < 2 σy, ou, P+Q < 3 Sa, onde Sa é a tensão admissível, de forma a
garantir-se a permanência da estrutura no regime elástico, evitando-se assim com que
hajam plastificações sucessivas. [12] Por outro lado, se P+Q ≥ 2 σy, será imposto um
regime de deformações plásticas sucessivas a nível da estrutura como um todo, que
podem acarretar perdas de funcionalidade por distorção severa, ou, ao impor grandes
pertubações no campo de tensões reinante devido essa mesma distorção, induzir a
nucleação de trincas e daí a ruptura da parede; é o "colapso incremental" (″ratchetting″),
fenômeno ao qual os tambores encontram-se particularmente sujeitos. Figura 3.13. Tal
nível de deformação deu-se após aproximadamente 4 anos de operação e o reator sofrer
cerca de 1450 ciclos de coqueamento-descoqueamento.
Figura 3.13: Perfil do casco de um tambor de coque fabricado em aço 1Cr-1/2Mo
deformado radialmente. (Regap, MG)

73 Observa-se ainda que se a exigência P+Q < 2 σy for atendida, falhas por fadiga-
térmica ainda podem ocorrer, isto é, sem que um regime de plastificações sucessivas de
″grandes proporções″ se estabeleça.
A natureza cíclica de um carregamento termo-mecânico, aliado a diferença entre as
propriedades mecânicas do metal de solda (cordões circunferenciais de ligação dos
aneis), e do metal base, este último possuidor de resistência ligeiramente inferior, assim
deformando-se mais facilmente, faz com que o escoamento tenda predominar em
posições adjacentes aos cordões. Neste sentido, taxas de crescimento radial tão elevadas
quanto ∼8mm/ano tem sido registradas, [55], como representado esquematicamente na
Figura 3.14.
Figura 3.14: Representação esquemática de um tambor de coque cujo casco é composto
por seis aneis numa condição inicial, não deformado, e já apresentando
um avançado nível de deformação [57].
A seguir, na Figura 3.15, ilustra-se a ocorrência de fadiga-térmica em um anel de
distribuição de vapor de uma unidade de craqueamento catalítico, em tubo API 5L-B/3″.
Observa-se que o trincamento não é associado à deformações de grandes extensões e
que o arranjo das trincas circunferenciais se faz de forma ″quase″ paralela, característica
das trincas nucleadas por esse mecanismo.

74
Figura 3.15: Ilustração da ocorrência de fadiga-térmica em um anel de distribuição de
vapor. (Regap, MG)
3.1.2.3 Grafitização e a esferoidização da cementita
Temperaturas iguais ou superiores a 420ºC correspondem a condições sob as quais a
decomposição da cementita com o tempo deve ser esperada, Fe3C → 3 Fe + Cgrafita.
Como se indica na reação, essa decomposição se dá através da formação de nódulos de
grafita; é a ″grafitização″ aludida anteriormente.
Na medida em que a resistência mecânica da grafita é, relativamente ao aço, muito
baixa, a grafitização pode se constituir em um evento extremamente grave se os nódulos
se agruparem. É o que tende a ocorrer na ZTA de juntas soldadas, nas regiões
submetidas à temperaturas próximas a 750ºC por decorrência da imposição do ciclo
térmico de soldagem. [20]
É ainda possível a esferoidização da cementita, inicialmente lamelar, ou,
simbolicamente, (Fe3C)lamelar → (Fe3C)globular, ocorrência de muito menor gravidade por
acarretar apenas uma ligeira queda da resistência mecânica do aço. Como indica o
gráfico abaixo, ambos os fenômenos competem entre si. Por possuir uma configuração
de maior estabilidade ou de menor energia superficial, a cementita globular não deve se
degenerar em grafita, num intervalo de tempo que seja industrialmente significativo, tal
como ocorre com a cementita de morfologia lamelar, Figuras 3.16 a e b. [58] Na Figura
3.16(a), a tubulação, com 30″, foi construída a partir de chapas de aço A-515Gr55 e
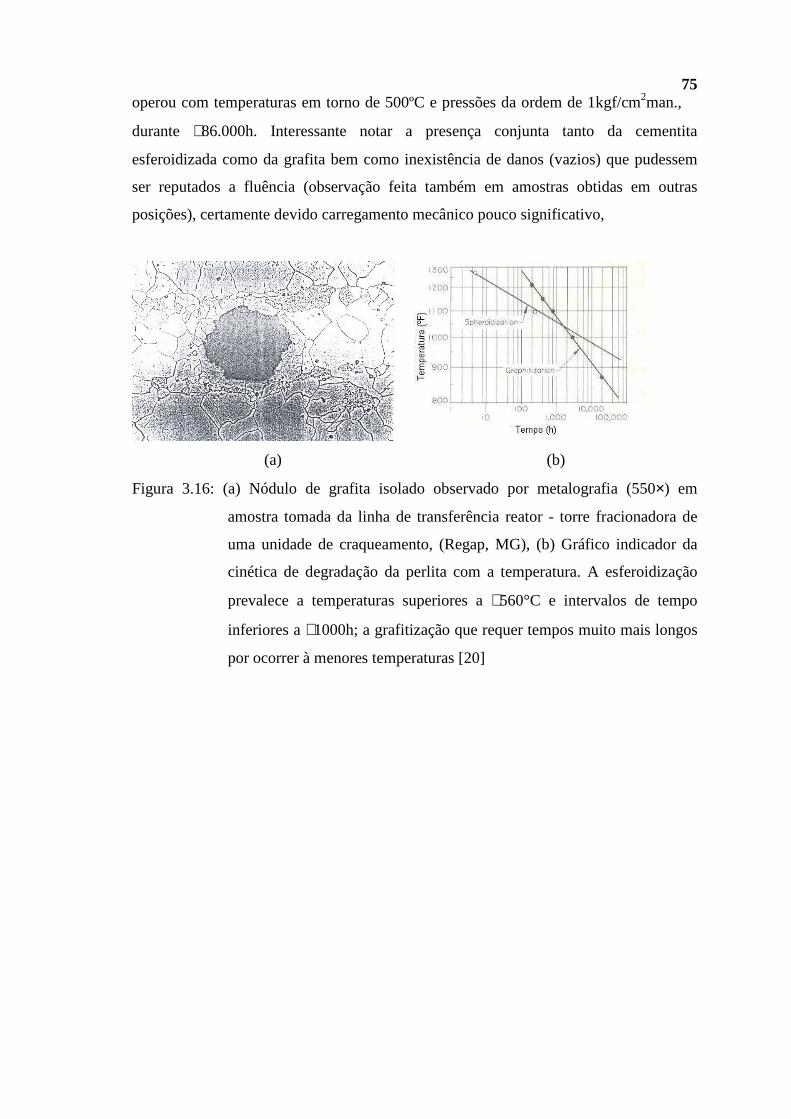
75 operou com temperaturas em torno de 500ºC e pressões da ordem de 1kgf/cm2man.,
durante ∼86.000h. Interessante notar a presença conjunta tanto da cementita
esferoidizada como da grafita bem como inexistência de danos (vazios) que pudessem
ser reputados a fluência (observação feita também em amostras obtidas em outras
posições), certamente devido carregamento mecânico pouco significativo,
(a) (b)
Figura 3.16: (a) Nódulo de grafita isolado observado por metalografia (550×) em
amostra tomada da linha de transferência reator - torre fracionadora de
uma unidade de craqueamento, (Regap, MG), (b) Gráfico indicador da
cinética de degradação da perlita com a temperatura. A esferoidização
prevalece a temperaturas superiores a ∼560°C e intervalos de tempo
inferiores a ∼1000h; a grafitização que requer tempos muito mais longos
por ocorrer à menores temperaturas [20]

76 3.1.2.4 Oxidação
Os meios gasosos são classificados em termos da atividade ou do potencial de oxigênio,
µO2, derivado do equilíbrio,
M + O2 = MO2, (2.24)
em que
µ Oo o o
2= - R.T.ln (k) = G = H T. S∆ ∆ ∆+ , (2.25)
onde ∆Hº indica a variação da entalpia padrão de formação do óxido, ∆Sº a variação da
entropia, ambos tabelados, k é a constante de equilíbrio da reação dada por,
k =a
a .PMO
M O
2
2
. (2.26)
Os a’s designam atividades (= 1, em fases condensadas puras), PO2é a pressão parcial
do oxigênio e ∆Go expressa a variação de energia livre para a reação direta a T. Assim,
pode-se obter,
µ O Oo
O
H
R.T
S
R
2 2 2
o o
= R.T.ln (P ) = G P e∆∆ ∆
⇒ =+
. (2.27)
Tendo sido fixado o sistema metal-óxido de interesse e a temperatura, essa atmosfera
será ″oxidante″ se a PO2for suficientemente elevada de modo a dar origem e estabilizar
a forma oxidada; será ″redutora″ em caso contrário. [53, 59, 60]
Servem como exemplo de atmosferas com tendência oxidante, o próprio ar ou os gases
que resultam da combustão realizada com excesso de ar (O2 + CO2), empregando-se
combustíveis ″limpos″ tais como o gás natural. Tenderão a ser redutores se a combustão
se processar com queima estequiométrica ou sub-estequiométrica de combustíveis

77 contendo um certo nível de contaminantes (por exemplo) a base do enxofre
(SO2+CO2+CO), tal como usualmente ocorre com os óleos combustíveis. [53]
Na presença de atmosferas contendo CO-CO2, pode-se empregar o equilíbrio:
CO + ½ O2 = CO2 (2.28)
Para cálculos do PO2 (≡ µ O2
) associada a uma dada razão (P
PCO
CO2
):
µ O
CO
CO O
o
2
2
2
12
= R.T.ln P
P .P= G - 282000 +86,7.T
=∆
⇒ ×
P
P= P e
CO
COO
- 282000
RT+
86,7
R2
2
12 (2.29)
Desta forma, tal como nos equilíbrios contendo apenas fases condensadas e oxigênio, é
possível conhecer as características dos meios gasosos contendo CO/CO2 a uma dada
temperatura T, relativamente a qualquer equilíbrio metal-óxido. Como será visto a
seguir, existem formas gráficas que permitem obter o pO2 ou a razão CO/CO2 associados
a um dado equilíbrio metal-óxido. São os diagramas de Ellin gham. Figura 3.17. [59]
A, por exemplo, 700°C, a energia livre de formação do Cr2O3 é inferior a de formação
do NiO. Deduz-se daí, que o primeiro é então um óxido possuidor de maior estabilidade
do que o último.
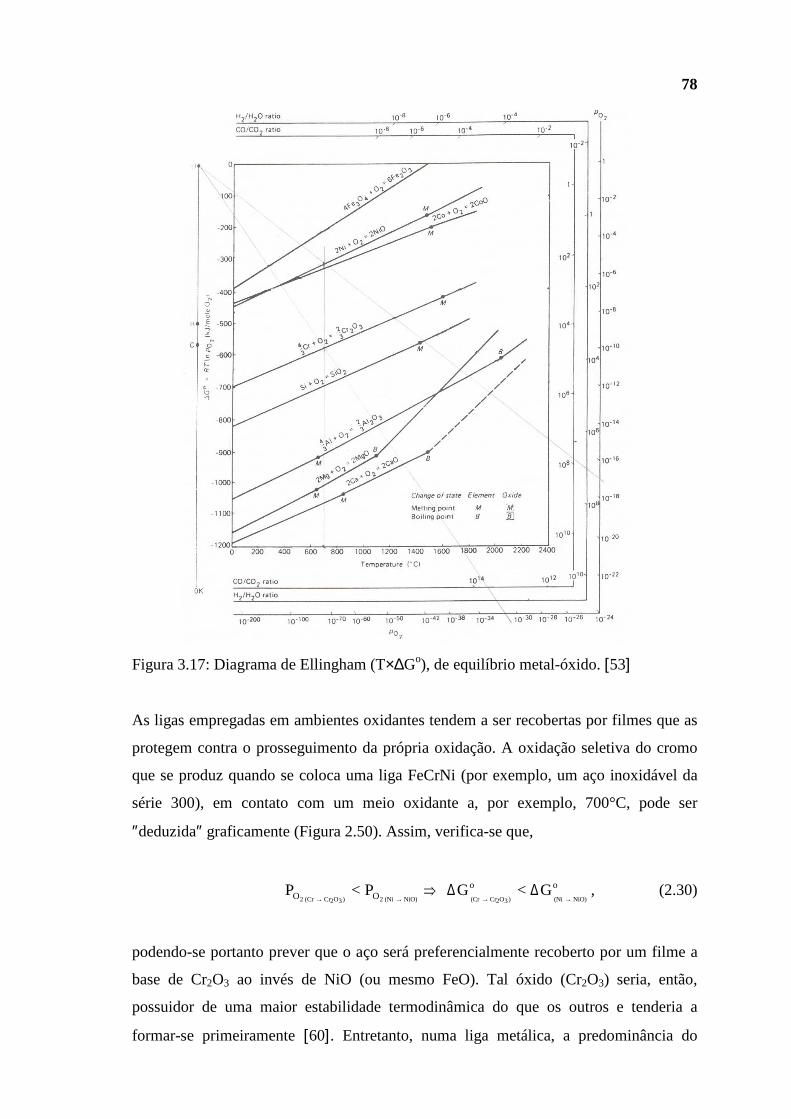
78
Figura 3.17: Diagrama de Ellingham (T×∆Go), de equilíbrio metal-óxido. [53]
As ligas empregadas em ambientes oxidantes tendem a ser recobertas por filmes que as
protegem contra o prosseguimento da própria oxidação. A oxidação seletiva do cromo
que se produz quando se coloca uma liga FeCrNi (por exemplo, um aço inoxidável da
série 300), em contato com um meio oxidante a, por exemplo, 700°C, pode ser
″deduzida″ graficamente (Figura 2.50). Assim, verifica-se que,
P < P G < GO Oo o
2 (Cr Cr2O3) 2 (Ni NiO) (Cr Cr2O3) (Ni NiO)→ → → →⇒ ∆ ∆ , (2.30)
podendo-se portanto prever que o aço será preferencialmente recoberto por um filme a
base de Cr2O3 ao invés de NiO (ou mesmo FeO). Tal óxido (Cr2O3) seria, então,
possuidor de uma maior estabilidade termodinâmica do que os outros e tenderia a
formar-se primeiramente [60]. Entretanto, numa liga metálica, a predominância do

79 óxido de uma natureza específica mostra-se dependente, também, da quantidade
relativa de cada metal na liga, Figura 3.18.
Figura 3.18: Evidência da ocorrência do óxido Cr2O3 com o aumento do teor de cromo,
o que leva ao aumento da resistência à oxidação de uma liga binária
FeCr. [53]
O gráfico coloca em evidência que as máximas resistências à oxidação que se pode
obter de ligas FeCr são conseguidas ao ter-se a presença majoritária do óxido de cromo
III, Cr2O3, o que ocorre a partir de cerca de 20% em peso. Mostra, também, que
aumentos no teor de cromo acarretam sensíveis aumentos da resistência a oxidação da
liga. Tais resultados foram obtidos ao colocar as amostras em um ambiente aquecido a
1000°C em uma atmosfera contendo 0,13 atm de O2. [53]
Pode também ser observado que uma liga Fe-16Cr (∼AISI 430) possui filmes contendo
misturas compostas pelo óxido Cr2O3 e pelo espinélio FeCr2O4, além da presença dos
óxidos de ferro, Fe3O4 e Fe2O3. Em uma liga Fe-9Cr, o óxido Fe2O3 ainda ocorre em
maior extensão ao passo que o Fe-0Cr (∼aço carbono) é revestido por uma mistura de
óxidos de ferro: a wustita (FeO, que se forma apenas acima de 570°C), a magnetita (um
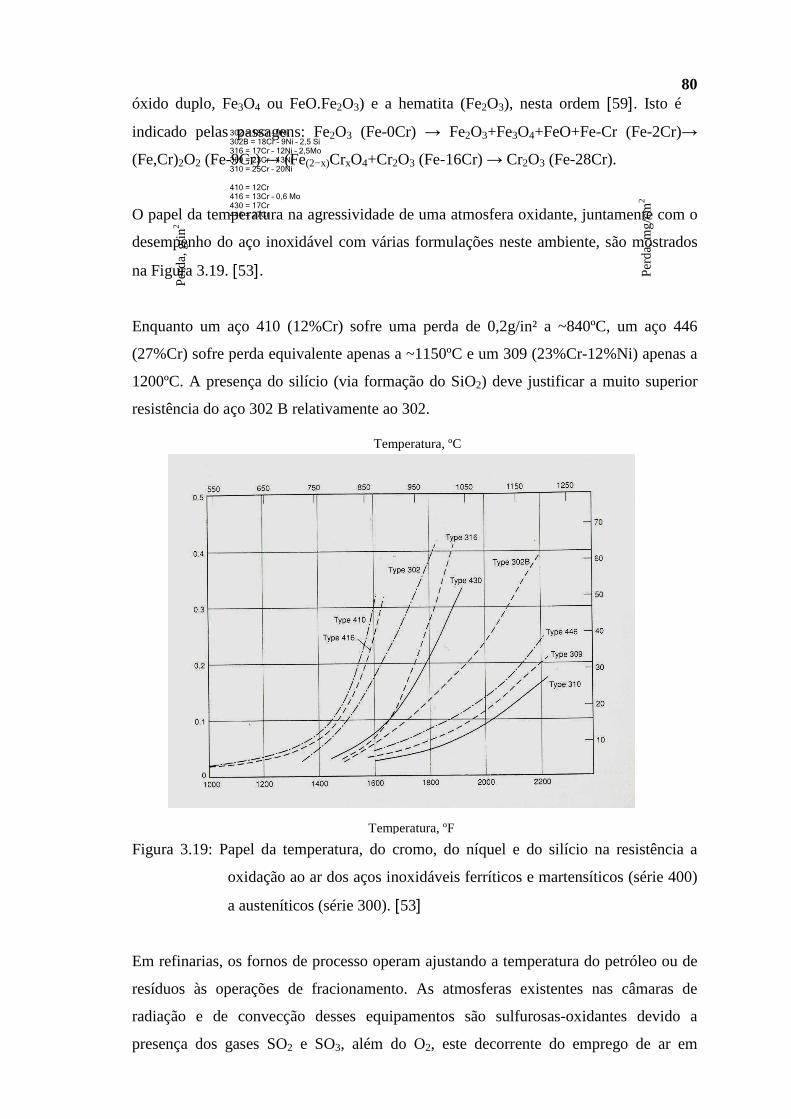
80 óxido duplo, Fe3O4 ou FeO.Fe2O3) e a hematita (Fe2O3), nesta ordem [59]. Isto é
indicado pelas passagens: Fe2O3 (Fe-0Cr) → Fe2O3+Fe3O4+FeO+Fe-Cr (Fe-2Cr)→
(Fe,Cr)2O2 (Fe-9Cr) → (Fe(2−x)CrxO4+Cr2O3 (Fe-16Cr) → Cr2O3 (Fe-28Cr).
O papel da temperatura na agressividade de uma atmosfera oxidante, juntamente com o
desempenho do aço inoxidável com várias formulações neste ambiente, são mostrados
na Figura 3.19. [53].
Enquanto um aço 410 (12%Cr) sofre uma perda de 0,2g/in² a ~840ºC, um aço 446
(27%Cr) sofre perda equivalente apenas a ~1150ºC e um 309 (23%Cr-12%Ni) apenas a
1200ºC. A presença do silício (via formação do SiO2) deve justificar a muito superior
resistência do aço 302 B relativamente ao 302.
Figura 3.19: Papel da temperatura, do cromo, do níquel e do silício na resistência a
oxidação ao ar dos aços inoxidáveis ferríticos e martensíticos (série 400)
a austeníticos (série 300). [53]
Em refinarias, os fornos de processo operam ajustando a temperatura do petróleo ou de
resíduos às operações de fracionamento. As atmosferas existentes nas câmaras de
radiação e de convecção desses equipamentos são sulfurosas-oxidantes devido a
presença dos gases SO2 e SO3, além do O2, este decorrente do emprego de ar em
Per
da,
mg
/cm2
Temperatura, ºC
Per
da,
g/in2
Temperatura, ºF
302 = 18Cr – 9Ni
302B = 18Cr – 9Ni – 2,5 Si
316 = 17Cr – 12Ni – 2,5Mo
309 = 23Cr – 13Ni
310 = 25Cr – 20Ni
410 = 12Cr
416 = 13Cr – 0,6 Mo
430 = 17Cr
446 = 27Cr

81 excesso para a queima de combustíveis contendo como contaminantes, entre outros,
metais e compostos sulfurados.
Muito frequentemente, as serpentinas desses equipamentos são fabricadas em aço liga
do tipo Cr−Mo, e tem sua resistência à corrosão por oxidação, com a temperatura,
função do teor de cromo. Assim, por exemplo, enquanto um aço 2,25Cr-1Mo tem como
limite máximo de temperatura para exposição contínua nesses ambientes o valor de
580°C, um aço 9Cr-1Mo tem como limite 620°C. [43]
Entretanto, o petróleo e suas frações (particularmente as pesadas) são susceptíveis de
degradarem-se termicamente junto à parede dos tubos, formando um sólido a ele
aderido (coque). Cargas contendo altos teores de asfaltenos (resíduos) são
particularmente susceptíveis.
A degradação térmica do poetróleo pode se dar sempre que houver incidência direta de
chama em uma dada região da superfície externa dos tubos por danificação do
queimador (usualmente ocorre por erosão do bico injetor), e descontrole da geometria
da chama. Neste caso, formam-se bolsões de vapor de hidrocarboneto os quais impedem
que haja uma refrigeração eficiente do tubo naqueles pontos. São, então, criadas as
condições apropriadas para que haja a degradação do hidrocarboneto.
Considerando que o coque possui um coeficiente de condutividade térmica que é de
apenas cerca de 1/50 da do aço e que a temperatura da câmara é da ordem de 850°C
(radiação), sobre-aquecimentos localizados relativamente às regiões adjacentes menos
aquecidas decorrerão dessa formação. Por conseqüência, além da redução da resistência
mecânica do aço (via queda da rigidez e da esferoidização da cementita), serão
intensificados tanto a cinética de oxidação externa como o acúmulo de danos por
fluência, em que o componente mecânico decorre da pressurização do tubo. Altas taxas
de oxidação acarretam a perda localizada da espessura de parede: Todos esses fatores
juntamente com a fluência, acarretam deformações localizadas (″laranjas″) que devem
prosseguir até a ruptura do tubo se a operação do forno não for interrompida em
antecedência. Nota-se que essa evolução se faz independentemente do tempo de
operação prévio do tubo. Figuras 3.20 a e b. Na Figura 3.20 são evidenciados os efeitos
da interposição de material pouco condutor de calor (coque) entre o fluido refrigerante
(petróleo) e a parede do tubo. Notar que o coque se forma apenas numa região, no

82 trecho de tubo voltado para a chama. Nessa região associam-se uma importante
redução de parede e espessa camada de óxidos.
Figura 3.20: (a) Ocorrência de ″laranja″ em um tubo A200-T5, 6″, componente da
serpentina de um forno de uma unidade de destilação atmosférica e, (b)
Seção reta do tubo em ponto coincidente com a região deformada.
(Regap, MG)
3.1.2.5 Descarbonetação e a carbonetação do aço. A carbonetação catastrófica
Um grande número de máquinas e equipamentos empregados em refinarias, como por
exemplo os eixos das bombas centrífugas e as molas helicoidais das válvulas de
segurança e alívio, dependem de componentes que são projetados para operar em
regime de fadiga. Por outro lado, a exposição dos aços à atmosferas cuja atividade do
carbono possibilita a alteração do teor de carbono superficial em serviço ou durante, por
exemplo, tratamentos térmicos ou termo-mecânicos, pode ocasionar importantes
alterações das propriedades superficiais devido a ocorrência da descarbonetação. É,
então, possível ter-se importantes modificações nas propriedades mecânicas nestas
regiões e, entre elas, o limite de fadiga, que é a tensão ou carregamento para o qual uma
peça de aço não deve romper por fadiga independentemente do número de ciclos de
cargas alternadas sobre ele aplicado.
São exemplos de peças que podem romper por fadiga os componentes mecânicos
fabricados em aço cujo serviço possa envolver carregamentos cíclicos em torção, como
são as molas referidas acima (de fato, não se espera que haja um grande número de

83 disparos ou de aberturas desse tipo de válvula em operação ou durante calibrações),
ou cíclicos em flexão, como é o caso dos eixos das bombas, cujo serviço seguramente
envolve um grande número de rotações. Uma vez que, em ambos os casos, a solicitação
máxima ocorre na ″fibra″ mais externa, tais componentes têm seu desempenho
condicionado à preservação da rugosidade, isso implicando que o aço deve possuir
adequada resistência a corrosão no meio sob consideração, e à adequação das
propriedades mecânicas superficiais, especificamente o limite de resistência (e assim a
dureza superficial), propriedade que guarda correlação direta com o limite de fadiga.
[61]
No que diz respeito ao comportamento dos aços a fadiga em alto ciclo (>103 ciclos),
considerar um aço SAE 1060 (%C=0,60), material de fabricação de um componente
mecânico que irá operar submetido à carregamentos alternados, como as molas
helicoidais. À fabricação dessas peças associa-se uma seqüência de operações
envolvendo altas temperaturas. Parte-se de barras previamente recozidas e a
conformação é presumidamente realizada “a quente”, isto é, acima da temperatura de
recristalização do aço. Para o ajuste das propriedades mecânicas requeridas pelo
serviço, emprega-se a têmpera seguida do revenido da peça, operação cujo sucesso é
criticamente dependente do teor de carbono.
Em quaisquer das etapas do processo em que a peça for submetida à temperaturas
superiores a cerca de 800°C, na qual o carbono apresenta-se totalmente solubilizado na
rede do aço, a ocorrência de descarbonetação (ou de carbonetação) deve ser
considerada. Na foto abaixo, Figura 3.21, mostra-se um aço SAE 1060 descarbonetado
(regiões claras próximas à superfície), ao ser mantido a 1200°C por 1h. Notar a
presença de uma fina camada de óxido na superfície. A região descarbonetada se
intensifica à medida em que se aproxima da superfície recoberta por um fino filme de
óxido.

84
Figura 3.21: Micrografia de um aço SAE 1060 exposto à temperatura de 1200°C por 1h.
Aumento: 50×. [59]
Entretanto, independentemente das características da atmosfera, estando o aço recoberto
por filmes de óxidos, o processo teria início através da reação entre o óxido na interface
óxido-aço com o carbono solubilizado na matriz austenítica (isto é, estando o aço
aquecido à temperaturas superiores a ∼800ºC), FeO + C → Fe + CO, reação que se
passa favoravelmente do ponto de vista termodinâmico pois, ∆Gº ≅ - 58kJ (a 1100°C),
constituindo-se a passagem C → CO, na “força motriz” para que a difusão do carbono
continue a se processar das regiões mais internas até a interface, dando seqüência ao
processo de descarbonetação.
Então, será necessário o escape do gás CO para que a descarbonetação tenha
continuidade. Dada as características do óxido que é gerado sobre a superfície do aço
carbono, usualmente contendo poros e fissuras, esse escape de fato é esperado ocorrer,
como apresentado na Figura 3.22.

85
Figura 3.22: Representação esquemática do mecanismo pelo qual a descarbonetação de
um aço oxidado deve se desenvolver. [adapt., 59]
Um aspecto importante deste processo é a avaliação da severidade da descarbonetação à
profundidade x, contada a partir da interface metal-óxido, decorrido um intervalo de
tempo t em que a peça se encontra aquecida à temperatura T.
A equação que modela a variação do teor de carbono na profundidade x, cx, por
decorrência de processos difusionais, é dada por:
(2.31)
para D não dependente de cx. Essa equação têm por solução a função,
c -c
c - c= 1-erf
x
2 D.tx o
s o
, (2.32)
quando se estabelece como condição inicial cx=c0, em que c0 designa o teor de carbono
do aço em t=0 (0 < x < ∞), e como condição de contorno cx=cs, em que cs designa o teor
de carbono superficial, ou seja, em x = 0 para 0 < t < ∞, e ainda, cx = co quando x → ∞
(para todo t). Na expressão acima, D é o coeficiente de difusão do carbono na austenita,
dado por
D = 24,6 e- 17540
T×
(mm2/s), (2.33)

86
e erf é a “função-erro” cujo valor pode ser obtido diretamente de tabelas ou aplicando-se
a série:
( )erf x =2
x -x
3 1!
x
5 2!
3 5
π×
×+
×−
... , (2.34)
sendo erf(0) = 0 e erf(∞) = 1. [62, 63]
Então, uma peça de aço inicialmente com 0,6%C terá após, por exemplo, 3h a 1100°C a
1,5 mm de profundidade o teor de carbono reduzido para cerca de 0,45%
(c -0,6
0-0,6= 1-erf
1,5
2 10x
-4 × ×
180 60=0,45%). Ao se fazer variar a profundidade x,
pode-se obter um perfil que expressa a redução do teor de carbono no aço (cx × x), a
qual será similar ao perfil esquemático dado na Figura 3.23.
Figura 3.23: Perfil ilustrativo da variação da composição do carbono a partir da
interface aço-óxido. Na figura, cs<co e cs→co com x. [adapt., 59]
Além do papel do óxido em um processo de descarbonetação, a interação do meio
gasoso com o metal, também viabilizado pela existência de fissuras e de poros
existentes no óxido, pode levar ao fenômeno inverso, a carbonetação.

87
Devido a presença do aço em atmosferas contendo misturas CO+CO2 em temperatura
elevada em refinarias, condição essa presente nos regeneradores das UFCC′s, há o
interesse em poder antecipar as possíveis conseqüências. Considerando o equilíbrio
expresso pela reação de Bouduard, 2 CO = CO2 + C, com ∆Gº = −170.550 + 174,3 T,
duas serão as possibilidades: carbonetação se a atividade do carbono ac no meio e no
metal apresentarem ac (meio) > ac (metal), descarbonetação se ac (meio) < ac (metal).
[53]
Repetindo o procedimento empregado anteriormente, parte-se da expressão, ∆Gº = −
RT lnk, onde k é a constante de equilíbrio da reação dada por, k =a a
P
c P
CO2
CO2×
, com P
representando pressões parciais dos gases envolvidos. Fazendo as substituições obtém-
se,
− 170.550 + 174,3T = 8,31T ln(k =a a
P
c P
CO2
CO2×
) (2.35)
∴ ac ≅ P
PeCO
2
CO
20606
T-21
2
×
, (2.36)
em que ac expressa a atividade do carbono no meio gasoso [59]. Assim, a (por exemplo)
800°C, ac ≅ 1,66×10−1 P
PCO2
CO2
, e a 1000ºC, ac ≅ 8×10−3 P
PCO2
CO2
. Então, para P
PCO
CO2
constante, verifica-se que um aumento da temperatura leva a um decréscimo da
atividade do carbono no meio.
Essa tendência pode ser vista no gráfico apresentado na Figura 3.24, onde a atividade do
carbono é colocada em função da razão (P
PCO2
CO2
) a várias temperaturas. A esse conjunto
de dados foram superpostas as atividades do carbono de alguns aços. Observa-se que,
nos aços inoxidáveis austeníticos, o carbono apresenta atividades (∼10−2)
significativamente inferior à aquela apresentada no aço carbono (>1) e no aço liga
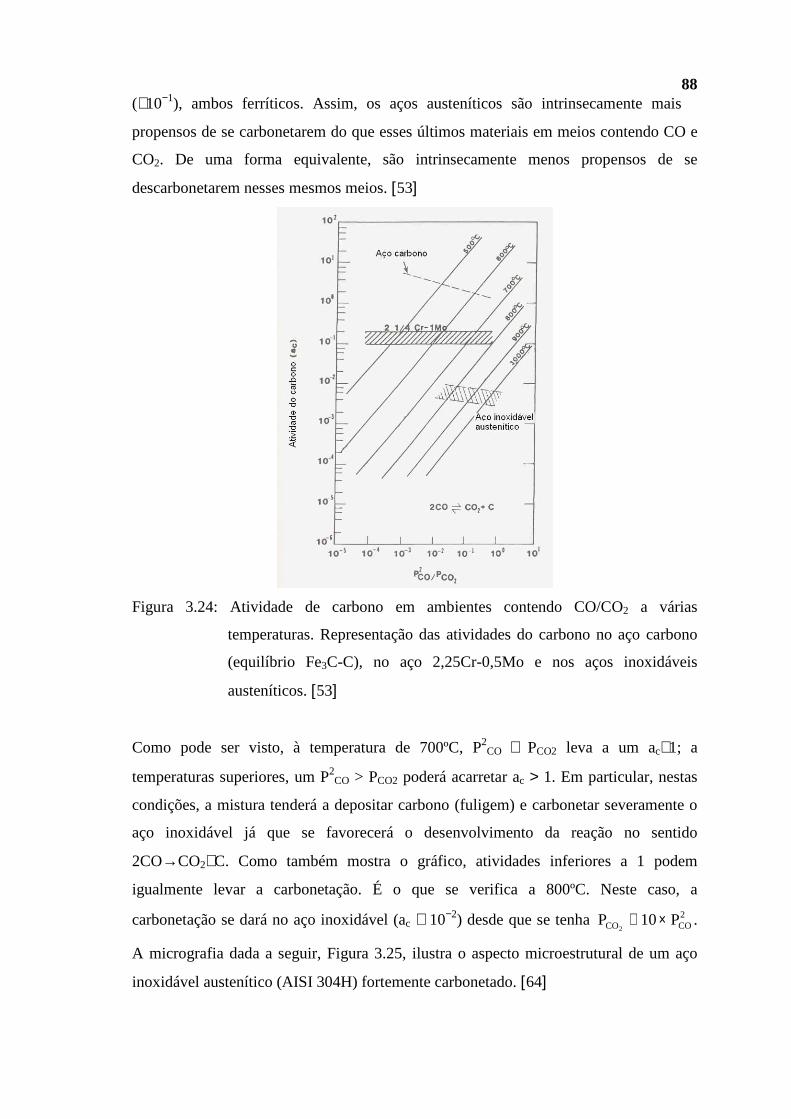
88 (∼10−1), ambos ferríticos. Assim, os aços austeníticos são intrinsecamente mais
propensos de se carbonetarem do que esses últimos materiais em meios contendo CO e
CO2. De uma forma equivalente, são intrinsecamente menos propensos de se
descarbonetarem nesses mesmos meios. [53]
Figura 3.24: Atividade de carbono em ambientes contendo CO/CO2 a várias
temperaturas. Representação das atividades do carbono no aço carbono
(equilíbrio Fe3C-C), no aço 2,25Cr-0,5Mo e nos aços inoxidáveis
austeníticos. [53]
Como pode ser visto, à temperatura de 700ºC, P2CO ≅ PCO2 leva a um ac≅1; a
temperaturas superiores, um P2CO > PCO2 poderá acarretar ac > 1. Em particular, nestas
condições, a mistura tenderá a depositar carbono (fuligem) e carbonetar severamente o
aço inoxidável já que se favorecerá o desenvolvimento da reação no sentido
2CO→CO2+C. Como também mostra o gráfico, atividades inferiores a 1 podem
igualmente levar a carbonetação. É o que se verifica a 800ºC. Neste caso, a
carbonetação se dará no aço inoxidável (ac ≅ 10−2) desde que se tenha P PCO CO2
2≅ ×10 .
A micrografia dada a seguir, Figura 3.25, ilustra o aspecto microestrutural de um aço
inoxidável austenítico (AISI 304H) fortemente carbonetado. [64]

89
Figura 3.25: Micrografia de um aço AISI 304H obtido de amostra da câmara plena de
um regenerador de uma UFCC. O aço apresenta-se fortemente carbonetado
após exposição por cerca de 15 anos a uma mistura CO-CO2 (CO2>CO) a
∼700°C. (Regap, MG)
Foi observado que a exposição de uma liga FeCrNi em ambiente oxidante acarreta a
reação: Cr → Cr2O3. O óxido de cromo é um óxido de características refratárias e
responsável pela adequação dessas ligas ao serviço a altas temperaturas em atmosferas
oxidantes. Entretanto, por conseqüência da oxidação do cromo, as camadas ″sub-
superficiais″ tornam-se empobrecidas neste elemento. É, então, necessário que o cromo
nas ligas resistentes ao calor seja formulado de modo que, no caso da ruptura do óxido,
esse seja prontamente reconstituído, de forma a manter inalterada a resistência a
oxidação ou ao calor da liga. Isso é conseguido ajustando o teor de cromo a um mínimo
de 20% (ver Figura 2.51). Dado que a carbonetação pode acarretar uma importante
redução do cromo solubilizado numa matriz austenítica (ou ferrítica) devido a afinidade
Cr-C, a resistência a oxidação a altas temperaturas da liga pode vir a comprometer-se de
forma irreversível. Tal ocorre pelo fato de que, atingido o limite de solubilidade em
solução sólida do carbono na matriz, este deve precipitar-se sob a forma dos carbonetos;
Cr23C6, Cr7C3 ou Cr3C2, a depender dos teores, da atividade do carbono na liga e da
temperatura, reduzindo-se o cromo, elemento responsável pela formação e reformação
do óxido, Figura 3.26. [65].

90
Figura 3.26: Diagrama de estabilidade termodinâmica dos elementos cromo e silício
para os sistemas Cr-O-C e Si-O-C obtidos a 1000 ºC. [65]
A carbonetação catastrófica (″metal dusting′′), é um fenômeno que se desenvolve nas
ligas resistentes ao calor, e entre elas, o aço inoxidável, quando tais ligas fazem contato
com meios gasosos com atividade ac > 1, usualmente entre 400 e 900°C. Ocorre,
inicialmente, carbonetando o metal, ou seja, primeiramente faz-se necessário a
formação de carbonetos, que se desintegram em grafita e particulado metálico,
formando uma mistura pulverulenta que, juntamente com fragmentos de óxidos,
caracteriza seu produto de corrosão. A etapa de desintegração pode ser acompanhada
pela ejeção desse particulado, processo que será favorecido se a superfície em questão
estiver fazendo contato com gases em escoamento. Como resultado, podem formar-se
alvéolos ou depressões na superfície metálica. [66]
Essas condições parecem terem sidas satisfeitas no espaço ou ″gap″ existente entre a
″malha hexagonal de ancoragem″ (AISI 304H) do concreto refratário anti-erosivo do
tubo vertical (″riser″) do regenerador de uma UFCC. Em serviço, esse conjunto faz
contínuo contato com misturas em fluxo ascendente contendo CO, CO2 e catalisador

91 (CO2>CO) a cerca de 750°C, devendo proteger as paredes desse tubo da ação erosiva
dos catalisadores (Al2O3), Figuras 3.27 a e b.
Figura 3.27: (a) Foto ilustrativa da malha (geometria hexagonal), AISI 304H, e do
concreto anti-erosivo fixos na parede do ″riser″ do regenerador tal como se
encontrava após uma campanha de cerca de 5 anos e, (b) Malha corroída
por carbonetação catastrófica; à sua esquerda mostra-se uma outra em
muito melhores condições. (Regap, MG)
3.1.2.6 Sulfetação e o ataque por cinzas fundidas
De forma análoga à abordagem anterior, um ambiente contendo compostos sulfurosos
será oxidante se o potencial de oxigênio associado for suficientemente alto de forma a
manter os óxidos estáveis. Será redutor se tal não ocorrer, estabilizando-se sulfetos em
lugar de óxidos (será alto o potencial de enxofre, µS2, neste caso). [53]
Ambos os ambientes podem ser gerados pela queima de hidrocarbonetos nos fornos de
processo. Se a queima for realizada com excesso de ar, formam-se os gases SO2 e SO3 e
o ambiente externo às serpentinas tenderá a ser oxidante. No caso de uma queima feita
com insuficiência de ar (o que não ocorre nestes casos), o enxofre tenderá ser
convertido em H2S, formando-se um ambiente redutor, em princípio, mais agressivo do
que um outro oxidante (uma vez que, tipicamente, óxidos conferem proteção superior
do que sulfetos). Em resumo, havendo o prevalecimento de uma atmosfera redutora, a
sulfetação predominará à oxidação. [53]
Entretanto, como foi apontado anteriormente, em refinarias se manuseia
hidrocarbonetos contendo quantidades variáveis de compostos sulfurados orgânicos e

92 particularmente o H2S. Tais ambientes serão, portanto, redutores e tenderão a
favorecer a formação de sulfetos como, por exemplo, CrS, e não de óxidos como, por
exemplo, o Cr2O3. A Figura 3.28 apresenta o diagrama de estabilidade termodinâmica
Cr-S-O. Ambientes encontrados em refinarias envolvendo hidrocarbonetos e o H2S
devem acarretar µS2 >> µO2, o que deve favorecer a formação e estabilização do CrS nas
ligas FeCrNi em detrimento do Cr2O3.
Figura 3.28: Diagrama esquemático de estabilidade termodinâmica. Sistema Cr-S-O.
[53]
Misturas puramente gasosas ou bifásicas (gás-líquido) contendo hidrocarboneto e o H2S
circulam a várias temperaturas, em vários pontos da planta. Dada a possibilidade da
degradação do óleo, a temperatura das correntes contendo hidrocarbonetos são
usualmente limitadas em cerca de 400°C. Nestas condições, a corrosão promovida pelo
sulfeto ao aço carbono, aço liga e inoxidável (<20%Cr) levará a formação de filmes em
que o FeS ainda deve predominar, Fe + H2S → FeS + H2, atingindo-se um máximo de
agressividade em cerca de 380ºC. Especificamente, a intensidade desse processo
corrosivo será, para uma dada metalurgia, função da temperatura e do teor do H2S
presente no meio. As taxas de corrosão resultantes da exposição do aço a esses
ambientes são apresentadas graficamente sob a forma das ″curvas McConomy″
apresentadas na Figura 3.29.
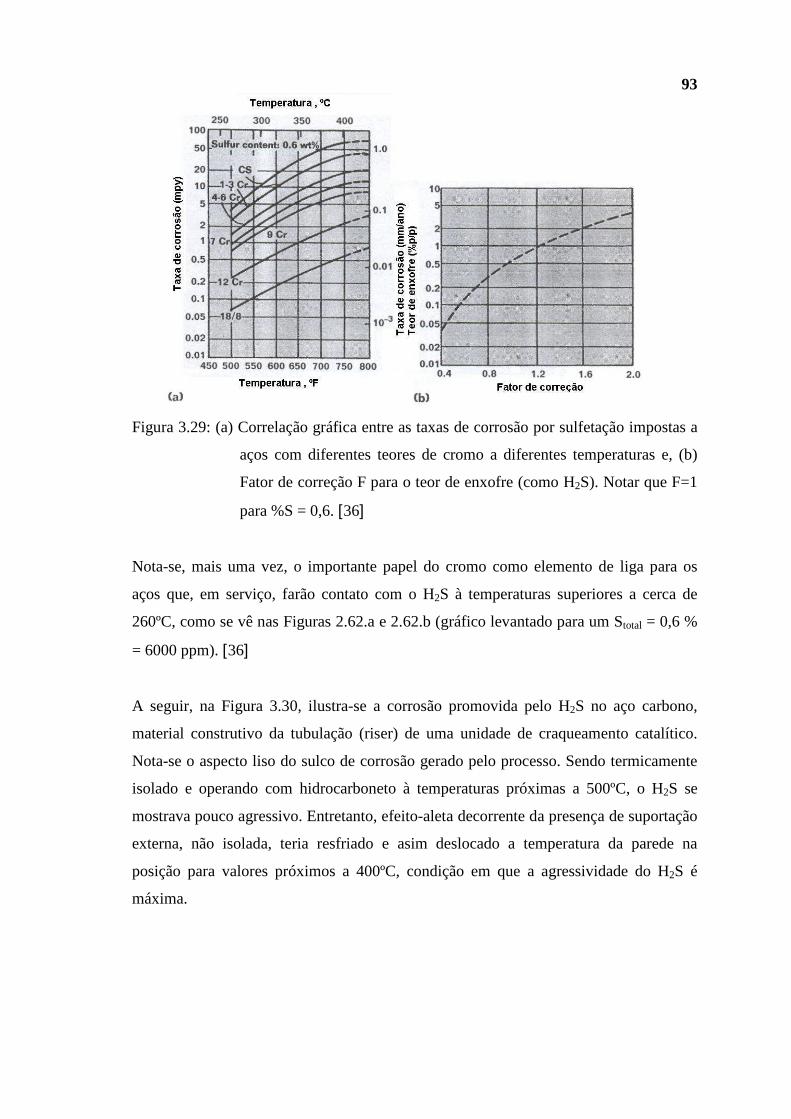
93
Figura 3.29: (a) Correlação gráfica entre as taxas de corrosão por sulfetação impostas a
aços com diferentes teores de cromo a diferentes temperaturas e, (b)
Fator de correção F para o teor de enxofre (como H2S). Notar que F=1
para %S = 0,6. [36]
Nota-se, mais uma vez, o importante papel do cromo como elemento de liga para os
aços que, em serviço, farão contato com o H2S à temperaturas superiores a cerca de
260ºC, como se vê nas Figuras 2.62.a e 2.62.b (gráfico levantado para um Stotal = 0,6 %
= 6000 ppm). [36]
A seguir, na Figura 3.30, ilustra-se a corrosão promovida pelo H2S no aço carbono,
material construtivo da tubulação (riser) de uma unidade de craqueamento catalítico.
Nota-se o aspecto liso do sulco de corrosão gerado pelo processo. Sendo termicamente
isolado e operando com hidrocarboneto à temperaturas próximas a 500ºC, o H2S se
mostrava pouco agressivo. Entretanto, efeito-aleta decorrente da presença de suportação
externa, não isolada, teria resfriado e asim deslocado a temperatura da parede na
posição para valores próximos a 400ºC, condição em que a agressividade do H2S é
máxima.
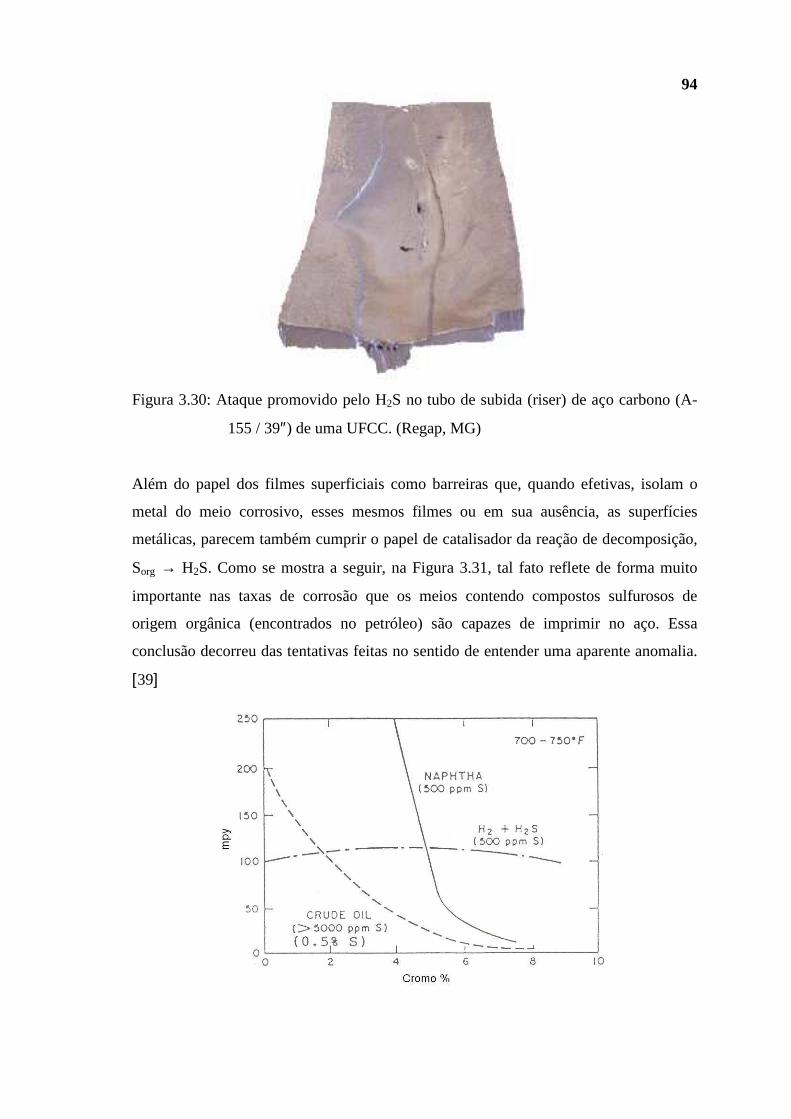
94
Figura 3.30: Ataque promovido pelo H2S no tubo de subida (riser) de aço carbono (A-
155 / 39″) de uma UFCC. (Regap, MG)
Além do papel dos filmes superficiais como barreiras que, quando efetivas, isolam o
metal do meio corrosivo, esses mesmos filmes ou em sua ausência, as superfícies
metálicas, parecem também cumprir o papel de catalisador da reação de decomposição,
Sorg → H2S. Como se mostra a seguir, na Figura 3.31, tal fato reflete de forma muito
importante nas taxas de corrosão que os meios contendo compostos sulfurosos de
origem orgânica (encontrados no petróleo) são capazes de imprimir no aço. Essa
conclusão decorreu das tentativas feitas no sentido de entender uma aparente anomalia.
[39]

95 Figura 3.31: Taxas de corrosão (mpy) apresentadas por aços contendo teores
variáveis de Cr como elemento de liga (0 a 8%), quando se encontravam
expostos a nafta + 500 ppmSorg, ao petróleo + 5000 ppm Sorg e ao H2 +
500 ppmH2S, à temperaturas entre 370 a 400ºC. [39]
A partir de uma série de experimentos, concluiu-se que a reação Sorg → H2S é catalisada
pelas superfícies com as quais os compostos sulfurosos fazem contato, sendo efetiva
(como catalisador) nos aços com até ∼5%Cr. Tal fato explicaria tanto a importante
queda das taxas de corrosão nos aços com teores superiores a ∼7%Cr, caso em que o
″H2S-catalítico″ (petróleo/nafta+Sorg → ... + H2S), deve responder pela corrosão, como a
constância das taxas quando o composto agressivo é o H2S previamente presente (não
catalítico e associado ao H2). Além disso, concluiu-se também que o H2S-catalítico seria
mais agressivo do que o não catalítico pelo simples fato de já ser formado junto às
paredes metálicas [39]. Por sua vez, a decomposição térmica das frações mais pesadas
do petróleo sobre a superfície metálica (a nafta é menos ″efetiva″ neste particular),
″desativando-a″ e protegendo-a, responderia pelas menores taxas observadas neste
último caso (petróleo com 0,5%S), não obstante o teor muito maior de compostos
sulfurosos contido naquele meio.
Paralelamente, apenas escoamentos à velocidades superiores a cerca de 30m/s
acarretariam elevação das taxas de corrosão [39]. Tal se daria devido ao transporte mais
efetivo do H2S do ″bulk″ até a interface com a parede metálica, o que seria
acompanhado por um efeito mecânico mais importante (tensões cisalhantes) de remoção
dos filmes protetores e, possivelmente, erosão.
O óleo combustível queimado nos fornos de processo normalmente contém, além dos
compostos sulfurados, quantidades importantes de sódio (como NaCl solubilizado na
água emulsionada no óleo) e de compostos organo-metálicos contendo níquel e vanádio
principalmente, que se segregam nos cortes mais pesados. Além da formação de uma
atmosfera sulfurosa-oxidante, a combustão desses materiais leva à formação de
″cinzas″. As cinzas são constituídas por particulado sólido a base de sulfatos e
vanadatos, que terminam por depositar-se sobre todas as superfícies presentes nas
câmaras de radiação e convecção desses fornos.

96 Enquanto permanecerem sólidas, permanecerão inócuas aos materiais sobre os quais
se depositam. Entretanto, ao se liqüefazerem darão origem a eletrólitos altamente
agressivos às ligas ferrosas existentes nesses locais. A corrosão assim promovida é
então denominada corrosão ou ataque por "cinzas fundidas". (″fuel ash corrosion″). [53]
Alguns dos componentes dessas cinzas podem possuir pontos de fusão bastante
inferiores à temperatura da câmara de radiação de um forno de processo, ∼850°C. É o
caso do V2O5 (que funde a 691ºC) ou da mistura eutética ∼90V2O5.∼10Na2SO4 (que
funde a ∼500ºC). [53]
O ataque se desenvolveria mediante a ″escorificação″ do óxido que recobre a liga ou
aço em questão, ao dar origem a um meio líquido onde se favorece termodinamicamente
sua incorporação. Por exemplo, a interação entre o sal Na2SO4 líquido (que funde a
885ºC) com o Cr2O3 sólido, óxido presente na superfície dos aços inoxidáveis
(>20%Cr), acarretará a escorificação desse último; literalmente, a dissolução do óxido
na cinza liqüefeita, destruindo-o. Esse processo pode desenvolver-se por um mecanismo
de ″escorificação básico″, Cr2O3 + O2 →2 CrO22−, em que o óxido dissolve-se como um
cromato e no qual o potencial de oxigênio na interface metal-cinza é alto o suficiente,
ou, alternativamente, por ″escorificação ácida″, Cr2O3 + SO42−→2Cr3+ + 3O2− + SO2 +
O2, no qual o óxido, ao reagir com o sulfato se dissolve sob a forma do cátion Cr3+ e do
ânion O2−. [59]
Presumindo que a escória líquida permaneça retida sobre a superfície e que seja
permeável aos compostos gasosos presentes no meio, o resultado seria a exposição de
uma liga despassivada a um ambiente a que, inicialmente, era capaz de resistir. Na
Figura 3.32, observa-se que enquanto o suporte encontra-se completamente destruído
por ação das cinzas, o trecho de tubo adjacente está preservado embora tenha,
evidentemente, estado em serviço submetido ao mesmo ambiente que levou a deposição
de cinzas no primeiro. Os níveis de temperatura no suporte foram suficientes para
liqüefazê-las ao passo que o tubo, por se encontrar abaixo do ponto de liqüefação
(∼550ºC), não.

97
Figura 3.32: Ataque por cinzas fundidas em um suporte, A-298GrHK-40, de um tubo da
câmara de radiação de um forno de uma unidade de destilação
atmosférica. (Regap, MG)
Problemas dessa natureza podem ser contornados por duas rotas: injeção de aditivos ao
óleo combustível (exemplo, Mg(OH)2), que acarretam a produção de cinzas com alto
ponto de fusão (por exemplo, o 3MgO.V2O5 funde a 1191°C), ou a seleção de ligas
resistentes tal como a 50Ni-50Cr. [53]

98 CAPÍTULO 4
4.1 Ataque pelo hidrogênio a altas temperaturas. A fragilização pelo revenido e por
precipitação de intermetálicos. O ataque intergranular por ácidos
politiônicos e a corrosão por sais de amônio
Em determinados trechos das unidades de hidrotratamento (HDT’s) associam-se ao
hidrocarboneto aquecido à temperaturas superiores a 200°C, misturas H2+H2S. Tal
como na presença do H2S, o aço pode sofrer corrosão por sulfetação, normalmente
uniforme, em que, mais uma vez, o H2S-catalítico deve cumprir importante papel.
Entretanto, são particularmente agressivos os vapores contendo apenas a mistura
H2+H2S. Tem-se verificado taxas de corrosão superiores em até 50% quando se tem por
referência a observada na presença do hidrocarboneto, [67], o qual pode se decompor e
proteger o aço ou, quando não decomposto, aderir e recobrir (ainda sob a forma líquida)
a parede metálica exercendo papel similar ao do coque.
Formam-se, nestes casos, ambientes fortemente redutores nos quais os sulfetos, e não os
óxidos, serão termodinamicamente estáveis. Os gráficos de Couper e Gorman,
envolvendo como variáveis a temperatura, o meio diluente, a concentração de H2S e as
taxas de corrosão resultantes, são largamente empregados na seleção dos aços e na
fixação das respectivas sobre-espessuras de corrosão.
Tendo a nafta como diluente, são mostrados dois desses gráficos, um para o aço
carbono e outro para um aço 12Cr, Figuras 4.1 a e b. Enquanto o aço carbono sofre
corrosão a uma taxa de 15mpy (0,38mm/ano) a 700ºF (371ºC) quando em contato com
0,1mol%H2S (340ppm ou 0,034%), o 12Cr tem essa taxa reduzida para 5mpy
(0,13mm/ano), ou reduzida em ∼66%, o que serve para ilustrar mais uma vez o papel do
cromo como elemento de liga do aço para o serviço nesses ambientes.[39]

99
Figura 4.1: (a) Taxas de corrosão (mpy) que decorrem do contato do aço carbono com
misturas H2 + H2S na nafta, tendo como variável a concentração de H2S (%mol) e a
temperatura (ºF) e, (b) idem para o aço 12Cr (AISI 405/410). [39]
Assim, diante de misturas H2S + H2, um mínimo de 12Cr é necessário para assegurar
reduções significativas das taxas de corrosão. [67]
Além do ataque corrosivo promovido pelo H2S ao aço, o H2 pode provocar sua
descarbonetação superficial. É o que tende ocorrer a ″altas temperaturas″ (>450ºC) e a
baixas pressões parciais de hidrogênio (<7MPa). Por outro lado, elevadas pressões
parciais e temperaturas não necessariamente altas, mas acima de cerca de 220°C, podem
favorecer a dissociação do na superfície (H2 → H + H) e, tendo-se o hidrogênio sob a
forma atômica, ocorre a absorção e a difusão do hidrogênio no aço [68]. Esse é um
fenômeno cujas conseqüências serão muito mais graves do que uma simples
descarbonetação superficial. Neste caso, o hidrogênio pode ocasionar a descarbonetação
interna (essencialmente a decomposição da cementita), e, subseqüentemente, o
trincamento e a fragilização do aço. O trincamento seria resultante da geração de

100 metano, o qual seria produzido a partir da reação de descarbonetação, Fe3C + 2 H2
→ 3 Fe + CH4. Não podendo difundir na rede cristalina do aço devido suas
(relativamente) grandes dimensões, o metano tende a acumular-se em interfaces como
são os contornos de grão. Produz-se, assim, um tensionamento interno originado da
pressurização que o gás acumulado acarreta na posição, o que termina por nuclear
trincas intergranulares. Figuras 4.2 a e b. [69]
Figura 4.2: (a) Micrografia de um aço carbono fissurado intergranularmente devido ao ataque pelo hidrogênio a altas temperaturas. Observar a descarbonetação da matriz nas adjacências da trinca. 25X. e, (b) Ampliação da foto à esquerda, 250X. [20]
Em resumo, a exposição do aço a ambientes contendo misturas gasosas H2S e H2 pode
acarretar tanto reduções de espessuras devido à sulfetação (ataque pelo H2S), como
fragilizações sob a forma do trincamento interno (ataque pelo H2), devendo ambos os
fatos serem observados na seleção do aço. [68, 69]
Assim, como é usual selecionar-se aços com base nos gráficos de Couper e Gorman
quando se tem o hidrocarboneto com misturas gasosas contendo H2S + H2, é usual
empregar-se as ″curvas de Nelson″ para a seleção do aço quando se tem caracterizado o
serviço com H2 independentemente da presença do H2S, Figura 4.3.
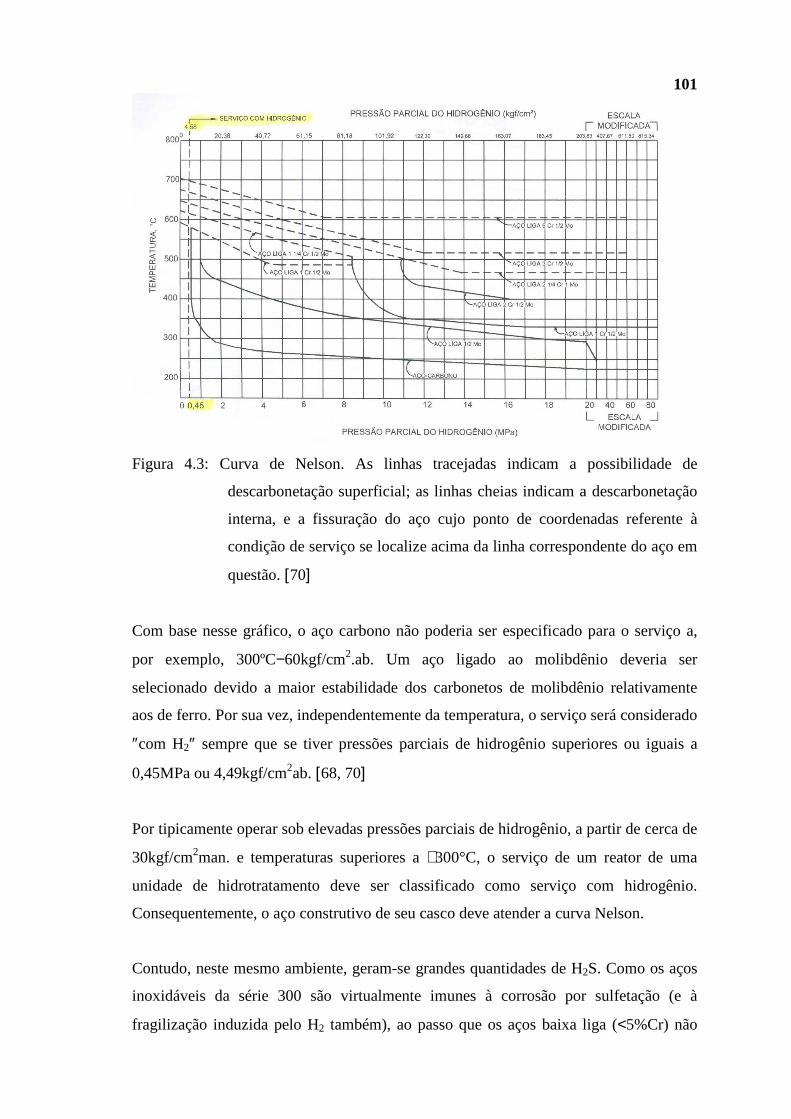
101
Figura 4.3: Curva de Nelson. As linhas tracejadas indicam a possibilidade de
descarbonetação superficial; as linhas cheias indicam a descarbonetação
interna, e a fissuração do aço cujo ponto de coordenadas referente à
condição de serviço se localize acima da linha correspondente do aço em
questão. [70]
Com base nesse gráfico, o aço carbono não poderia ser especificado para o serviço a,
por exemplo, 300ºC−60kgf/cm2.ab. Um aço ligado ao molibdênio deveria ser
selecionado devido a maior estabilidade dos carbonetos de molibdênio relativamente
aos de ferro. Por sua vez, independentemente da temperatura, o serviço será considerado
″com H2″ sempre que se tiver pressões parciais de hidrogênio superiores ou iguais a
0,45MPa ou 4,49kgf/cm2ab. [68, 70]
Por tipicamente operar sob elevadas pressões parciais de hidrogênio, a partir de cerca de
30kgf/cm2man. e temperaturas superiores a ∼300°C, o serviço de um reator de uma
unidade de hidrotratamento deve ser classificado como serviço com hidrogênio.
Consequentemente, o aço construtivo de seu casco deve atender a curva Nelson.
Contudo, neste mesmo ambiente, geram-se grandes quantidades de H2S. Como os aços
inoxidáveis da série 300 são virtualmente imunes à corrosão por sulfetação (e à
fragilização induzida pelo H2 também), ao passo que os aços baixa liga (<5%Cr) não

102 são imunes, esses reatores são construídos com aços ligados, que respondem pela
função estrutural, e são revestidos internamente com aço inoxidável (AISI 347), Figura
2.13.
Pelo fato das matrizes austeníticas oferecerem uma maior solubilidade ao hidrogênio do
que as ferríticas, cerca de uma ordem de grandeza superior, e também pelo fato da
difusividade deste elemento nas matrizes austeníticas ser muito inferior, cerca de duas
ordens de grandeza em uma ampla faixa de temperaturas, o revestimento interno tende
a reter a quase totalidade do hidrogênio absorvido. [69]
Entretanto, devido às reduções da solubilidade com o abaixamento da temperatura, parte
do hidrogênio dissolvido deve difundir-se na rede austenítica indo acumular-se nas
proximidades da interface com o substrato (um aço estrutural ligado ao Cr-Mo), haja
visto sua baixa solubilidade nos ferríticos. Tal fato é evidenciado no gráfico da Figura
4.4. Em operação (corresponde ao ″0h″), um reator pressurizado a 150kgf/cm2 e a
450ºC, apresenta uma concentração de equilíbrio de 40ppmH no revestimento, nas
proximidades da superfície livre em contato com o gás. Essa concentração se reduz até
cerca de 30ppm junto a interface, lado austenítico, contra apenas cerca de 3ppm, lado
ferrítico. Ao atingir a temperatura ambiente, após proceder-se um resfriamento a uma
taxa de 35,4ºC/h (12h), a concentração do hidrogênio na interface, lado ferrítico tende a
zero, ao passo que do lado austenítico se eleva até cerca de 110ppm; resfriando-se a
4,4ºC/h (96h), alcança-se uma concentração ligeiramente inferior, 100ppm, o que serve
para evidenciar o papel não muito significativo das taxas de resfriamento neste aspecto.
[71]

103
Figura 4.4: Perfis de distribuição do hidrogênio no revestimento (8 mm de espessura) e
no substrato (150 mm de espessura), em 3 condições: em operação (0h),
à temperatura ambiente após resfriar-se a uma taxa de 35,4ºC/h, e após
resfriar-se a uma taxa de 4,4ºC/h.[71]
No caso da obtenção do revestimento por soldagem, método largamente empregado
atualmente, relativamente ao de colaminação ou ao de explosão, considerações similares
às feitas acima se fazem necessárias por permitir selecionar adequadamente o
consumível que deverá fazer contato com o substrato. Isso porque devido aos efeitos da
diluição, é possível a formação de martensita na interface, justo do lado em que os
níveis de hidrogênio alcançam os maiores valores. Segue-se a possibilidade do
trincamento (FF) e do descolamento do revestimento, devido à pressurização exercida
pelo gás hidrogênio (H + H → H2) acumulado naquelas posições. Como resultado, é
usual o emprego de consumíveis do tipo 309L (baixo carbono) em paralelo com
recomendações de que não se pratique diluições superiores a 40%.
Os aços e os consumíveis de solda ligados ao Cr-Mo, quando mantidos à temperaturas
entre 325 e 575ºC [39], são também susceptíveis à ″fragilização pelo revenido″,
fenômeno que acarreta o aumento da temperatura de transição dúctil-frágil e a redução
do KIC do aço [71]. Portanto, este é um fenômeno ao qual os cascos soldados dos
reatores das plantas de hidrotratamento, Figura 4.5, em sua totalidade especificados em

104 aços deste tipo e, normalmente, submetidos à temperaturas superiores a 300ºC em
serviço, encontram-se sujeitos.
Figura 4.5: Casco de um reator de uma unidade HDT ainda na fábrica, atualmente em
operação. Metalurgia: A387Gr22 (21/2Cr-1Mo), normalizado-
revenido. (Regap, MG)
A fragilização pelo revenido ocorre apenas na presença de contaminantes específicos do
aço. Dentre eles, pode-se citar: o manganês, o fosfóro, o silício, o antimônio, o estanho
e o arsênio, os quais, com a temperatura, se segregam e são retidos nos contornos de
grão da austenita prévia, Figura 4.6. [48, 71]
Figura 4.6: Representação esquemática da austenita prévia em uma matriz ferrítica e em
uma matriz ferrito-perlítica. [48]

105 Dependendo da intensidade desta segregação, fixada a temperatura e o tamanho de
grão austenítico, diretamente dependente dos teores dos elementos contaminantes, pode-
se ter um significativo aumento da temperatura de transição e redução dos níveis de
energia necessários para causar a ruptura frágil do aço, tal como já mencionado. [48].
Diante desse fenômeno, deve-se procurar cumprir procedimentos de partida e paradas
do reator que evitem pressurizá-lo estando seu casco submetido a temperaturas
inferiores à de transição.
Considerando o emprego de placas de aço forjado (A387), a susceptibilidade de sua
fragilização em serviço é indicada pelo valor do ″fator J″ do aço,
J = 104 x (P+Sn) x (Si+Mn), (2.37)
em que as concentrações são dadas em %(p). Por sua vez, a susceptibilidade do metal de
solda é dada pelo valor do ″fator X″,
X =10 P + 5 Sb + 4 Sn + As
100 (2.38)
onde os teores são expressos em ppm [71]. O aço, seja na forma de placas laminadas ou
forjadas, bem como os respectivos metais de solda, serão considerados pouco
susceptíveis se apresentarem um J ≤ 100 e um fator X ≤ 12, respectivamente [71].
Talvez se possa justificar o emprego de diferentes expressões para inferir um mesmo
fenômeno ao se considerar as diferenças existentes entre as microestruturas de um aço
trabalhado termo-mecanicamente com aquela presente no metal de solda, considerando-
se, por exemplo, o papel da austenita prévia. [72].
De modo a determinar a propensão e a extensão da fragilização do aço e dos metais de
solda a serem empregados numa dada fabricação, foi proposto um tratamento térmico
que induzisse e acelerasse o fenômeno [73]. Trata-se do tratamento por resfriamento em
etapas (″step cooling″), Figura 4.7, que deve ser conduzido de forma contínua a cinco
diferentes níveis de temperatura e a diferentes tempos de encharque: 468/100, 496/60,
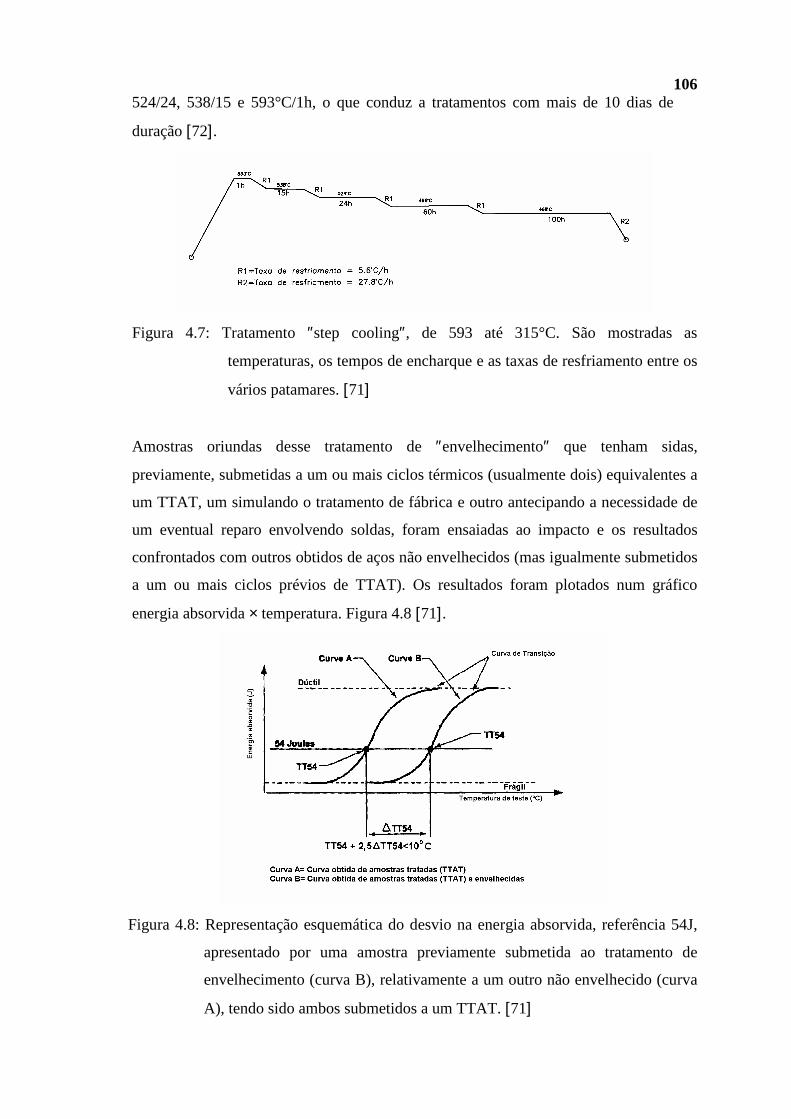
106 524/24, 538/15 e 593°C/1h, o que conduz a tratamentos com mais de 10 dias de
duração [72].
Figura 4.7: Tratamento ″step cooling″, de 593 até 315°C. São mostradas as
temperaturas, os tempos de encharque e as taxas de resfriamento entre os
vários patamares. [71]
Amostras oriundas desse tratamento de ″envelhecimento″ que tenham sidas,
previamente, submetidas a um ou mais ciclos térmicos (usualmente dois) equivalentes a
um TTAT, um simulando o tratamento de fábrica e outro antecipando a necessidade de
um eventual reparo envolvendo soldas, foram ensaiadas ao impacto e os resultados
confrontados com outros obtidos de aços não envelhecidos (mas igualmente submetidos
a um ou mais ciclos prévios de TTAT). Os resultados foram plotados num gráfico
energia absorvida × temperatura. Figura 4.8 [71].
Figura 4.8: Representação esquemática do desvio na energia absorvida, referência 54J,
apresentado por uma amostra previamente submetida ao tratamento de
envelhecimento (curva B), relativamente a um outro não envelhecido (curva
A), tendo sido ambos submetidos a um TTAT. [71]

107 Assim, para que se possa aplicar esse método, faz-se necessário levantar a curva de
transição nos dois casos. Cada curva seria plotada a partir de um número mínimo de
cinco pontos; cinco energias (média obtida a partir do ensaio de três corpos de prova),
obtidas em cinco temperaturas, sendo uma delas necessariamente −30ºC e outra que
assegurasse um comportamento dúctil, usualmente a própria temperatura ambiente. A
adequação do aço relativamente a uma baixa tendência de fragilizar-se em serviço, será
indicada pelo atendimento da desigualdade, TT54 + 2 ∆TT54 < 10ºC, onde TT54
representa a temperatura em que se deseja verificar a absorção de 54J de energia (obtida
por interpolação), para o grupo de amostras que não sofreu o envelhecimento, enquanto
∆TT54 = TT54 (E) − TT54, representa a variação ou o aumento da temperatura (a 54J)
para o grupo de amostras que foi submetido ao envelhecimento, TT54 (E), também
interpolada, relativamente ao que não o sofreu. [74]
O revestimento interno dos reatores encontram-se sujeitos a dois outros modos de falha.
O mais importante deles diz respeito a fragilização por fase intermetálica ″sigma″, fase
rica em cromo, Fe∼30%Cr, dura e intrínsecamente frágil. Esta fase afeta a tenacidade
(principalmente), a dureza, a ductilidade e mesmo as propriedades da liga relativamente
à corrosão localizada à temperatura ambiente.
Embora qualquer aço que contenha um mínimo de 16%Cr e um máximo de 32%Ni
possa desenvolvê-la por precipitação direta (eventualmente com os intermetálicos χ
(chi) e laves), desde que seja mantido por um período de tempo adequado entre 540 e
870ºC, no presente caso a fase seria formada através da decomposição da fase delta
(δ→σ), previamente presente nas microestruturas brutas de fusão em depósitos de
solda, a partir da qual a fase sigma se forma rapidamente, Figura 4.9. [41, 75]

108 Figura 4.9: Microestrutura evidenciando a presença de fase sigma, escura, acicular e
imersa na matriz austenítica de um aço AISI 304H. Componente (corpo)
de uma junta de expansão que em serviço permanecia em contacto com
gás a cerca de 750ºC. Aumento: 500×. (Regap, MG)
No reator, as regiões adjacentes a bocais e suportes e as ranhuras de posicionamento de
juntas de anéis (″o-rings″), isto é, todas aquelas posições que tendem a ser mais
solicitadas durante paradas, partidas e em serviço devido ao efeito concentrador de
tensões (k), estarão mais sujeitas ao trincamento por fragilização sigma. Esta é a razão
pela qual tais transições geométricas devem ser suavizadas ou ″adoçadas″. Se
k = 1+ 2b
ρ, expressa quantitativamente o efeito concentrador de um entalhe elíptico,
onde b representa a dimensão da elípse que é posicionada perpendicularmente à direção
de solicitação e ρ é o raio de curvatura orientado na direção de b, um adoçamento
corresponderá ao aumento de ρ e acarretará na redução de k. Além disto, sendo as
transições geométricas ″acabadas″ por soldagem manual (eletrodo revestido), é
procurado empregar-se um consumível que permita ajustar o teor da fase delta no
depósito à valores não superiores a ∼8%.
Por sua vez, os aços inoxidáveis ferríticos (≥12Cr) são particularmente propensos à
fragilização por α′ quando são mantidos aquecidos à temperaturas superiores a 340°C.
Figura 4.10. Tal processo é mais conhecido por ″fragilização a 475°C″, pelo fato de que
o seu efeito é maximizado nesta temperatura. Essa é também uma fase rica em cromo
que decorre da decomposição espinodal da ferrita α ou, Fe-α (ferro/cromo) → α (rica
em ferro) + α′ (rica em cromo), fases não distinguíveis por microscopia óptica. [41]

109 Figura 4.10: Prato (∅∼2,5m) em aço AISI 410s tal como se apresentava após cerca
de 5 anos de operação devido a fragilização α′. É posicionado no fundo de
uma torre fracionadora de uma UFCC. Em operação, encontrava-se
submetido à temperaturas próximas a 380°C. (Regap, MG)
Toda extensão do revestimento interno de um reator HDT encontra-se sujeito a sofrer o
ataque intergranular assistido por tensão (IGSCC) ou não (IGA), pelo ácido tetratiônico,
H2S4O6, o qual pode se formar durante paradas do equipamento ou da planta através da
interação ar úmido e sulfetos (produto de corrosão). Essa é a razão pela qual o aço
inoxidável selecionado é um aço estabilizado, por exemplo, ao Nb (AISI 347), uma vez
que apenas o aço sensitizado mostra-se susceptível. A necessidade de especificação do
aço estabilizado se verifica por duas razões: 1) a faixa de temperaturas empregadas para
o TTAT do aço Cr-Mo (substrato), 680 a 720ºC, tratamento que é realizado após a
obtenção do revestimento interno, coincide com aquela em que a cinética de
precipitação do carboneto Cr23C6 é particularmente favorecida, 2) as temperaturas
existentes em operação são, usualmente, superiores a 370ºC, temperatura apontada
(conservativamente) como limite inferior da faixa de tamperaturas (370-815ºC) na qual
a sensitização em serviço pode ocorrer. [41, 75, 76]
De modo a tornar a possibilidade desse ataque ainda mais remota, é recomendável
proceder a neutralização dessa superfície mediante aplicação de uma solução cáustica a
50ºBe (2%), ou a base de barrilha (Na2CO3 + H2O → 2 NaOH + CO2), de forma a obter
uma solução com pH ≥ 9 [76].
Além do H2S, geram-se também grandes quantidades de amônia nesses reatores. Dessa
forma, o hidrocarboneto dele efluente conterá H2 (injetado em excesso), NH3, H2S e
água (vapor). Sob essas condições, passa a existir a possibilidade da sublimação do sal
NH4HS, Figura 4.11 [77].
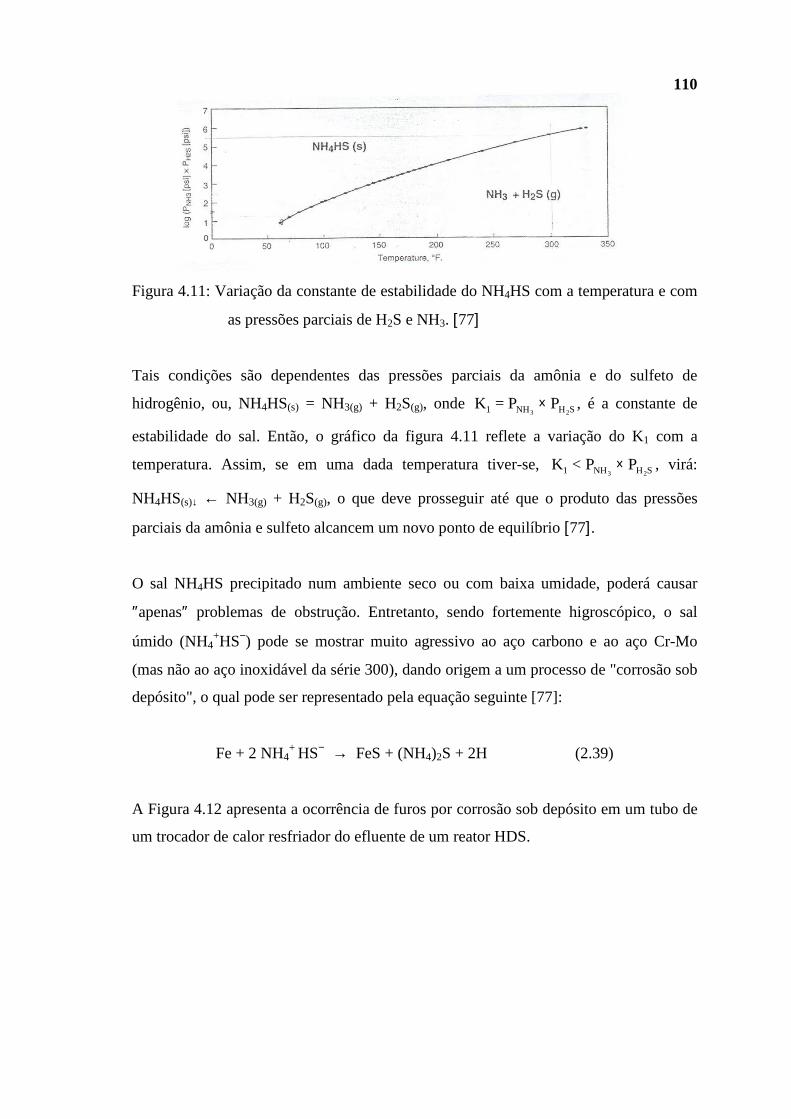
110
Figura 4.11: Variação da constante de estabilidade do NH4HS com a temperatura e com
as pressões parciais de H2S e NH3. [77]
Tais condições são dependentes das pressões parciais da amônia e do sulfeto de
hidrogênio, ou, NH4HS(s) = NH3(g) + H2S(g), onde K = P P1 NH H S3 2× , é a constante de
estabilidade do sal. Então, o gráfico da figura 4.11 reflete a variação do K1 com a
temperatura. Assim, se em uma dada temperatura tiver-se, K < P P1 NH H S3 2× , virá:
NH4HS(s)↓ ← NH3(g) + H2S(g), o que deve prosseguir até que o produto das pressões
parciais da amônia e sulfeto alcancem um novo ponto de equilíbrio [77].
O sal NH4HS precipitado num ambiente seco ou com baixa umidade, poderá causar
″apenas″ problemas de obstrução. Entretanto, sendo fortemente higroscópico, o sal
úmido (NH4+HS−) pode se mostrar muito agressivo ao aço carbono e ao aço Cr-Mo
(mas não ao aço inoxidável da série 300), dando origem a um processo de "corrosão sob
depósito", o qual pode ser representado pela equação seguinte [77]:
Fe + 2 NH4+ HS− → FeS + (NH4)2S + 2H (2.39)
A Figura 4.12 apresenta a ocorrência de furos por corrosão sob depósito em um tubo de
um trocador de calor resfriador do efluente de um reator HDS.

111
Figura 4.12: Ocorrência de furos por corrosão sob depósito em tubo 3/4″, A-209GrT1, de
um trocador de calor resfriador do efluente de um reator HDS; sendo o
fluido constituído de diesel, H2, NH3, H2S e vapor. Processo corrosivo
decorrente da presença de sal ácido no interior dos tubos. (Regap, MG)
É usual prevenir a formação deste sal mediante injeção de ″água de lavagem″. Essa
água deve ter limitados o teor de oxigênio (50ppb) e cloretos (50ppm), e deve,
evidentemente, ser injetada a montante do ponto em que são reunidas as condições de
pressão (pressões parciais) e de temperatura que possam levar à precipitação do sal.
Entretanto, soluções aquosas contendo esse sal podem também ser muito corrosivas.
Deve-se, assim, injetar água numa vazão que assegure uma concentração de bisulfeto
inferior a 2%(p) ou 0,04mol%. Do mesmo modo, em presença do sal, deve-se evitar
velocidades de escoamento que sejam superiores a cerca de 3m/s e assim a corrosão-
erosão, [39, 78]
Por razões distintas, tanto os sistemas de topo de destilações e de fracionadoras de
craqueamento catalítico, como as unidades de hidrotratamento, em suas seções de
reação, podem reunir condições que levem à formação e precipitação do sal NH4Cl:
NH4Cl(s) = NH3(g) + HCl(g) , K = P P2 NH HCl3× (2.40)
Tal como no caso anterior, o NH4Cl deve precipitar sempre que for atendida a
desigualdade, K < P P2 NH HCl3× . A representação gráfica do K2 é dada na Figura 4.13,
[77]
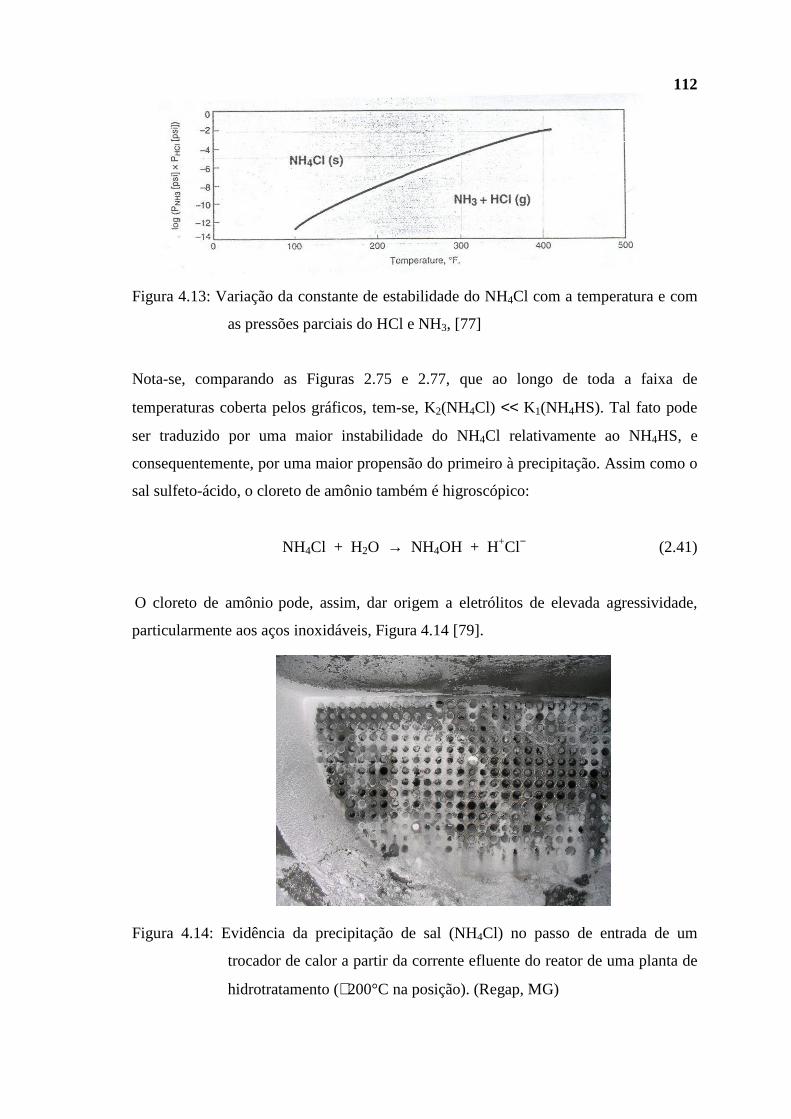
112
Figura 4.13: Variação da constante de estabilidade do NH4Cl com a temperatura e com
as pressões parciais do HCl e NH3, [77]
Nota-se, comparando as Figuras 2.75 e 2.77, que ao longo de toda a faixa de
temperaturas coberta pelos gráficos, tem-se, K2(NH4Cl) << K1(NH4HS). Tal fato pode
ser traduzido por uma maior instabilidade do NH4Cl relativamente ao NH4HS, e
consequentemente, por uma maior propensão do primeiro à precipitação. Assim como o
sal sulfeto-ácido, o cloreto de amônio também é higroscópico:
NH4Cl + H2O → NH4OH + H+Cl− (2.41)
O cloreto de amônio pode, assim, dar origem a eletrólitos de elevada agressividade,
particularmente aos aços inoxidáveis, Figura 4.14 [79].
Figura 4.14: Evidência da precipitação de sal (NH4Cl) no passo de entrada de um
trocador de calor a partir da corrente efluente do reator de uma planta de
hidrotratamento (∼200°C na posição). (Regap, MG)

113 CAPÍTULO 5
5.1 Corrosão naftênica
O ácido naftênico, um constituinte natural de determinados tipos de petróleo, incluindo
a maior parte daqueles oriundos da bacia de Campos no Brasil, diz respeito a toda uma
família de ácidos carboxílicos interligados por unidades CH2 à estruturas saturadas em
anel do tipo ciclo-alcano, usualmente um ciclo-pentano ou ciclo-hexano. Daí, a
denominação genérica ″ácidos naftênicos″. Podem, então, ser representados por
moléculas do tipo, R(CH2)mCOOH, m ≥ 1, onde R designa a estrutura em anel. Possuem
massa molar variando de cerca de 200 até cerca de 700g/mol, mas mostram tendência
de concentrar-se na faixa dos 400g/mol [9, 80, 81].
O nível de acidez do petróleo é geralmente expresso por um ″índice de acidez total″
(IAT), cujo valor é obtido por método titulométrico expresso em mgKOH/g óleo,
descrito na norma ASTM D664. Na medida em que esse índice resulta da contribuição
total dos vários outros grupamentos ácidos eventualmente presentes no hidrocarboneto
(por exemplo, ácidos alifáticos ou aromáticos ou mesmo ácidos minerais), não é
possível determinar uma acidez que seja relativa a um único constituinte numa base
molar. Tal limitação torna possível ter-se distintos petróleos e frações com um mesmo
IAT, mas possuidores de diferentes agressividades ao aço. Ainda assim, de uma forma
geral, um petróleo será considerado ″ácido″, e desta forma, um potencial causador de
problemas de corrosão naftênica se possuir um IAT ≥ 0,5mgKOH/g [82, 83], não
obstante existirem registros de casos de corrosão com IAT′s inferiores a 0,5 (casos
associados a hidrocarbonetos com baixos teores de enxofre e elevadas velocidades de
escoamento) [81, 84].
No que tange ao mecanismo de corrosão, verifica-se que o grupo carboxílico é o
responsável pela reatividade da molécula com as ligas metálicas, enquanto que as
demais estruturas respondem pela solubilidade em meio orgânico do sal que resulta do
processo corrosivo. Evidentemente, ambas as características cumprem importante papel
na agressividade desses compostos.

114 Tendo por referência o aço carbono e um ambiente contendo H2S, a corrosão
naftênica, que se desenvolve em meio exclusivamente líquido, se processaria segundo a
reação:
Fe + 2 RCOOH → Fe(RCOO)2 + H2, (2.42)
produzindo-se o naftenato de ferro que é solúvel no óleo. Em presença do H2S, o
naftenato, pouco volátil e viscoso, pode se regenerar segundo a reação:
Fe(RCOO)2 + H2S → FeS + 2RCOOH, (2.43)
podendo dar origem a um processo cíclico de corrosão-regeneração, Figura 5.1 [8, 82]:
2 RCOOH → Fe(RCOO)2 → 2 RCOOH... (2.44)
Figura 5.1: Ilustração do mecanismo de corrosão naftênica no qual o ácido é regenerado
através da reação naftenato x H2S, dando origem à cavidades
hemisféricas geometricamente bem definidas. [8]
Mecanismos de corrosão como o acima descrito seriam favorecidos sempre que o
contato metal-gota de óleo acidificada ocorra. Assim, a superfície dos componentes

115 metálicos horizontais internos de uma torre de destilação a vácuo ou atmosférica,
local em que tais gotículas podem condensar-se e permanecer em repouso, bem como os
recheios, estes devido a perda de carga que impõem ao fluxo ascendente de gases,
apresentam-se particularmente susceptíveis de sofrerem um ataque por este mecanismo
no qual o regime de escoamento parece não cumprir nenhum papel.
Um mecanismo de corrosão similar pode também ocorrer sobre superfícies verticais.
Diferem entre si pelo fato de que sobre uma superfície vertical um lento fluxo
descendente de misturas hidrocarboneto-naftenato (presumido lento devido a
viscosidade do naftenato e do óleo), tende ocorrer, neste caso podendo dar origem a
sulcos ou depressões oblongas em lugar de cavidades hemisféricas.
Assim, quanto mais lento for este escoamento, mais próximo se estará do processo
discutido para as superfícies horizontais, e mais parecido deverá ser o resultado do
processo corrosivo. Entretanto, como a experiência de campo indica, as paredes
(verticais) das torres tendem a ser menos atingidas pelo ácido. Além dos menores
″tempos de residência″, poder-se-ia argumentar que os componentes internos da torre
agem no sentido de convergir e manter o fluxo de gases na parte mais central do
equipamento, desfavorecendo o contato com as suas paredes.
Encontra-se com freqüência na literatura especializada a afirmativa de que para que haja
um efetivo aumento da resistência do aço inoxidável ao ataque pelos ácidos naftênicos,
faz-se necessário um teor de molibdênio igual ou superior a 2,5%. Assim, os aços AISI
316 (2 a 3%Mo), desde que com %Mo ≥ 2,5, ou AISI 317 (3 a 4%Mo), devem ser
selecionados para o serviço no qual a presença do ácido é esperada [8, 82, 83]. Os
resultados lançados no gráfico abaixo, Figura 5.2, corroboram essa afirmativa. [39]
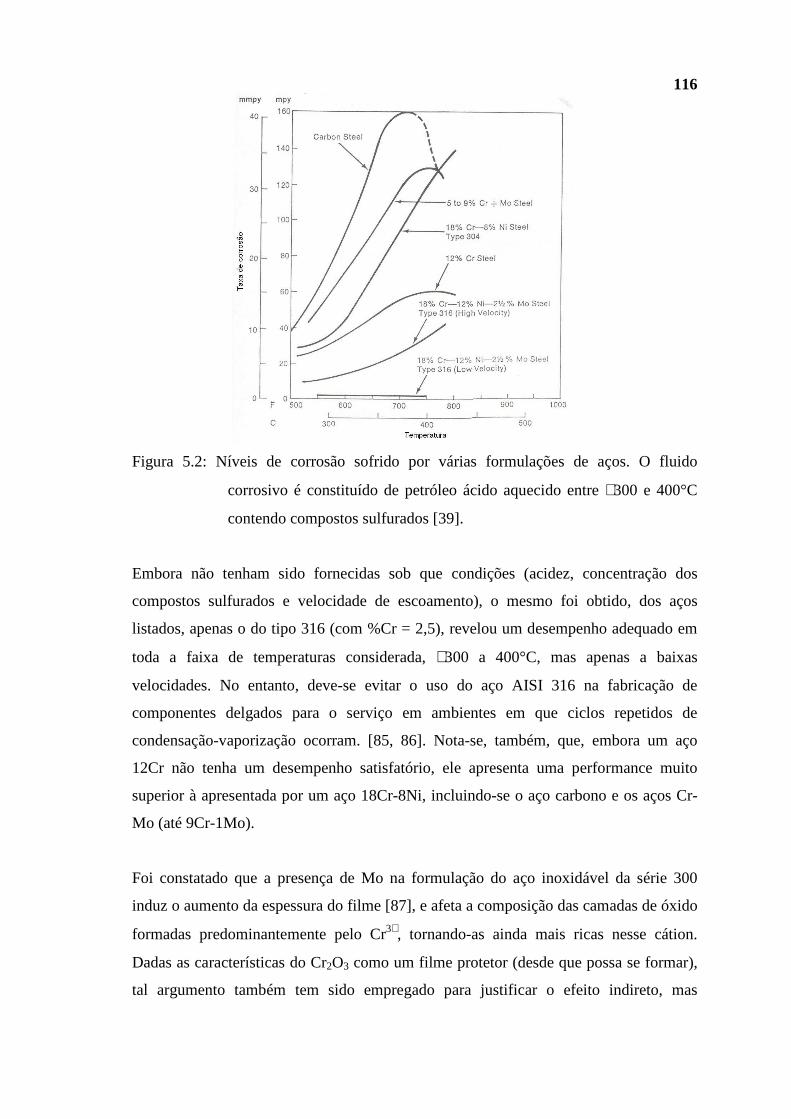
116
Figura 5.2: Níveis de corrosão sofrido por várias formulações de aços. O fluido
corrosivo é constituído de petróleo ácido aquecido entre ∼300 e 400°C
contendo compostos sulfurados [39].
Embora não tenham sido fornecidas sob que condições (acidez, concentração dos
compostos sulfurados e velocidade de escoamento), o mesmo foi obtido, dos aços
listados, apenas o do tipo 316 (com %Cr = 2,5), revelou um desempenho adequado em
toda a faixa de temperaturas considerada, ∼300 a 400°C, mas apenas a baixas
velocidades. No entanto, deve-se evitar o uso do aço AISI 316 na fabricação de
componentes delgados para o serviço em ambientes em que ciclos repetidos de
condensação-vaporização ocorram. [85, 86]. Nota-se, também, que, embora um aço
12Cr não tenha um desempenho satisfatório, ele apresenta uma performance muito
superior à apresentada por um aço 18Cr-8Ni, incluindo-se o aço carbono e os aços Cr-
Mo (até 9Cr-1Mo).
Foi constatado que a presença de Mo na formulação do aço inoxidável da série 300
induz o aumento da espessura do filme [87], e afeta a composição das camadas de óxido
formadas predominantemente pelo Cr3+, tornando-as ainda mais ricas nesse cátion.
Dadas as características do Cr2O3 como um filme protetor (desde que possa se formar),
tal argumento também tem sido empregado para justificar o efeito indireto, mas

117 extremamente benéfico do molibdênio, na resistência a corrosão naftênica
apresentada por essa classe de aços. [8]
É também possível encontrar na literatura a afirmativa de que os elementos de liga que
formam sulfetos estáveis envolvendo um maior número de átomos de enxofre, tais
como aqueles formados pelo cromo (Cr2S3 / FeCr2S4), e pelo molibdênio (MoS2), são
geradores de filmes superficiais mais densos e assim com maior poder protetor do que
aquele conferido unicamente pelo ferro (FeS). Tais formações tenderiam ocorrer
espontaneamente nos aços inoxidáveis imersos numa atmosfera com baixo potencial de
oxigênio, isto é, que fosse suficientemente rica em compostos sulfurados à temperaturas
intermediárias (da ordem de 400°C) [8].
Além da metalurgia, a severidade da corrosão naftênica é influenciada por outras
variáveis. Encontram-se citados: a temperatura, o IAT, o teor de H2S, o regime de
escoamento, a massa molar e a complexidade da molécula [81, 88].
O efeito da temperatura (Figura 5.2) pode ser avaliado de modo qualitativo ao se
considerar que a constante de velocidade k da reação de corrosão seja dada por uma
expressão do tipo Arrhenius [7], k ∝ e∆H/RT , onde ∆H é a energia de ativação, isto é,
aquela que os reagentes devem possuir para que efetivamente reajam. Nota-se, então,
que o aumento da temperatura T acarreta o aumento de k e, assim, agrava a severidade
do processo corrosivo, a qual verifica-se tornar-se importante apenas acima de cerca de
220°C, atingindo-se um máximo de agressividade a cerca de 380°C [82, 83]. À
temperaturas apenas pouco superiores a ∼400°C, o ácido passa a decompor-se, o que
ocorre em paralelo com o craqueamento do hidrocarboneto e a formação de coque (por
essa mesma razão, o H2S perde sua agressividade), o qual deve depositar-se sobre
filmes de sulfetos, reforçando-os. Ambos os fatos traduzem-se por uma redução da
severidade do ataque [80].
Como seria de se esperar, verifica-se que a agressividade do petróleo e de seus cortes
aumenta com o aumento da acidez, sendo que um dado corte tem sido apontado como
(potencialmente) agressivo se o seu IAT for igual ou superior a 1,5mgKOH/g. [83].
Como é possível verificar no gráfico apresentado na Figura 5.3, um óleo aquecido à
temperaturas apenas pouco superiores a 200°C com IAT de ∼2mgKOH/g, já seria capaz
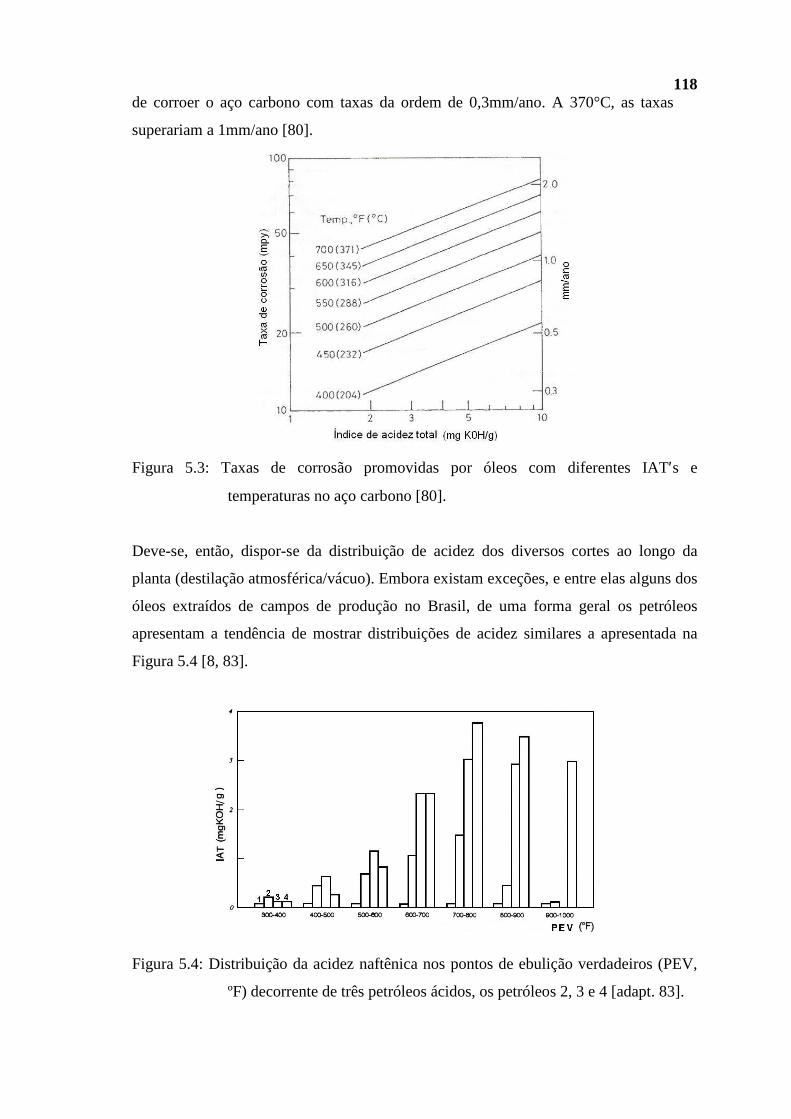
118 de corroer o aço carbono com taxas da ordem de 0,3mm/ano. A 370°C, as taxas
superariam a 1mm/ano [80].
Figura 5.3: Taxas de corrosão promovidas por óleos com diferentes IAT′s e
temperaturas no aço carbono [80].
Deve-se, então, dispor-se da distribuição de acidez dos diversos cortes ao longo da
planta (destilação atmosférica/vácuo). Embora existam exceções, e entre elas alguns dos
óleos extraídos de campos de produção no Brasil, de uma forma geral os petróleos
apresentam a tendência de mostrar distribuições de acidez similares a apresentada na
Figura 5.4 [8, 83].
Figura 5.4: Distribuição da acidez naftênica nos pontos de ebulição verdadeiros (PEV,
ºF) decorrente de três petróleos ácidos, os petróleos 2, 3 e 4 [adapt. 83].

119 Esta característica foi verificada existir a partir de quatro diferentes tipos de
petróleo, sendo apenas um deles considerado não ácido, o petróleo 1 (IAT’s não
fornecidos). Nos três casos em que se tinha petróleos com acidez dada como
significativa, verificou-se nítida tendência do ácido em maximizar-se nos cortes com
pontos de ebulição verdadeiro (PEV) entre 700 e 900°F (370 a 480°C). Pelo fato de que,
nestes níveis de temperaturas (>400°C), o hidrocarboneto já inicia sua decomposição,
ele deve ser destilado num ambiente com a pressão reduzida, como é o que ocorre numa
destilação (torre) a vácuo. Desta forma, nestas condições, estes cortes são de fato
encontrados a apenas a ∼390°C (∼20mmHg), fração correspondente ao dos gasóleos de
reciclo e pesado (GOR/GOP).
Consequentemente, os internos e costado da torre existentes na elevação correspondente
à do GOR-GOP são precisamente aqueles sujeitos a sofrer corrosão com maior
intensidade por um mecanismo que envolva a condensação de gotas ácidas, Figura 5.5.
Figura 5.5: Corrosão promovida por ácidos naftênicos no revestimento (AISI 410s) do
costado de uma torre de uma unidade de destilação a vácuo, elevação
GOR. (Regap, MG)
Como foi assinalado, o sal que resulta da corrosão naftênica sofrida pelo aço carbono,
aço liga ou aço inoxidável, (Mn+(RCOO)n) é solúvel no óleo. Assim, do ponto de vista
estrito do ataque naftênico ao aço, a variável ″velocidade de escoamento″ não deveria
assumir maior importância, pois como resultado da corrosão, não são gerados filmes
protetores que fossem susceptíveis de danificação por efeito mecânico. Entretanto, tanto
a taxa na qual o ácido é transportado do fluido para a interface metal-fluido, bem como

120 a taxa na qual o sal é removido da mesma interface, deverão se intensificar em
fluidos escoando a altas velocidades. Portanto, maiores deverão ser as taxas de corrosão
quando se tem fluidos escoando a velocidades elevadas, quando todos os demais fatores
interferentes se mantiverem iguais. Verificou-se haver uma redução de 50%
(12→6mm/ano) nas taxas de corrosão sofridas por curvas em aço carbono quando se
reduzia a velocidade do fluido de 64% (73→26m/s); para curvas em aço 5Cr-0,5Mo,
essa redução foi de 70% (2→0,6mm/ano) [89].
Além do mais, o H2S, em maior ou menor proporção, invariavelmente se faz presente
nos petróleos, e assim, nas suas frações. Consequentemente, deve-se sempre esperar a
presença do FeS, ou de sulfetos de natureza diversa, insolúveis no óleo, que possam
recobrir e (eventualmente) passivar o aço. Consequentemente, a remoção do filme
ocasionada por efeito mecânico resultante do escoamento (tensões cisalhantes) poderá
intensificar sobremaneira a corrosividade do fluido. Em particular, tanto o escoamento
turbulento como a turbulência localizada induzida por acidentes geométricos como as
reduções em tubulações ou raízes de cordões de solda (Figura 5.6), são apontados como
potenciais causadores de corrosão localizada severa. Como os fatos demonstram, é,
precisamente, o que pode ocorrer numa linha de transferência forno-torre de uma
destilação a vácuo [85].
Figura 5.6: Ilustração de como uma diminuta protuberância tal como a raiz de um
cordão de solda pode perturbar o escoamento na camada limite, alterar as
tensões cisalhantes que lá se desenvolvem, induzir turbulência localizada e
acarrretar um processo de corrosão-erosão à jusante da raiz [90].
O efeito da massa e da estrutura do ácido na corrosividade ao aço carbono foi
investigado experimentalmente. Os resultados mostraram claramente que, numa dada
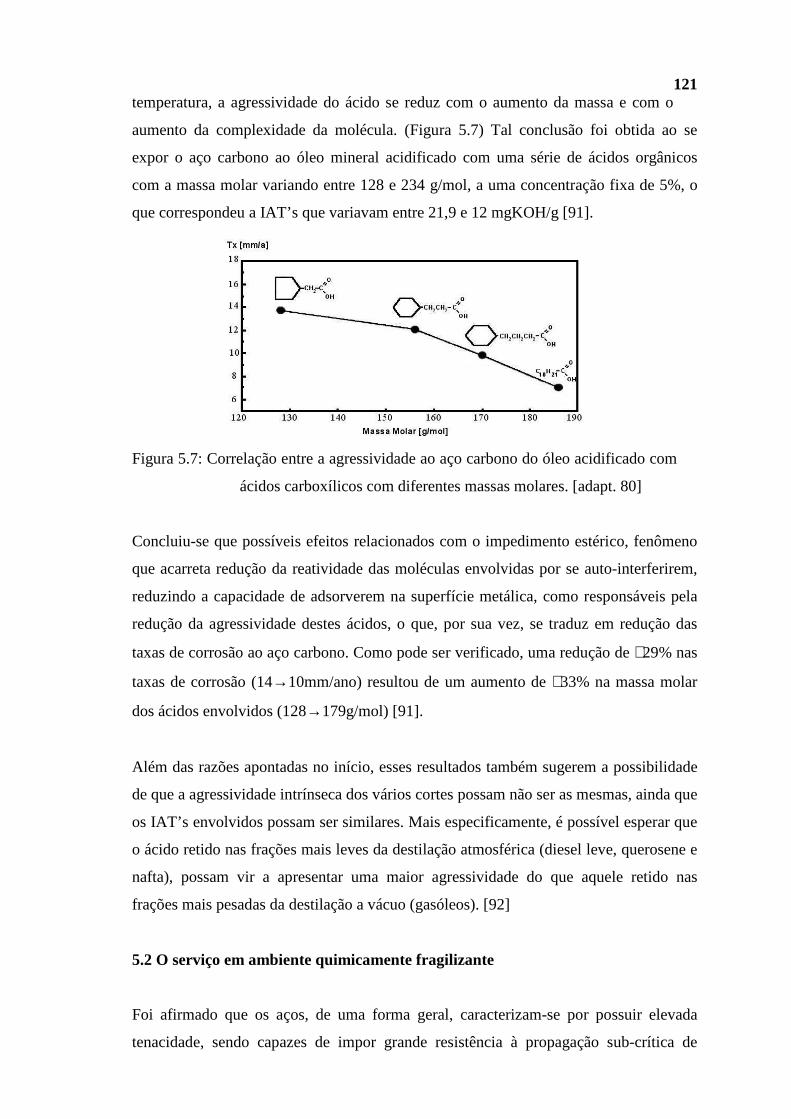
121 temperatura, a agressividade do ácido se reduz com o aumento da massa e com o
aumento da complexidade da molécula. (Figura 5.7) Tal conclusão foi obtida ao se
expor o aço carbono ao óleo mineral acidificado com uma série de ácidos orgânicos
com a massa molar variando entre 128 e 234 g/mol, a uma concentração fixa de 5%, o
que correspondeu a IAT’s que variavam entre 21,9 e 12 mgKOH/g [91].
Figura 5.7: Correlação entre a agressividade ao aço carbono do óleo acidificado com
ácidos carboxílicos com diferentes massas molares. [adapt. 80]
Concluiu-se que possíveis efeitos relacionados com o impedimento estérico, fenômeno
que acarreta redução da reatividade das moléculas envolvidas por se auto-interferirem,
reduzindo a capacidade de adsorverem na superfície metálica, como responsáveis pela
redução da agressividade destes ácidos, o que, por sua vez, se traduz em redução das
taxas de corrosão ao aço carbono. Como pode ser verificado, uma redução de ∼29% nas
taxas de corrosão (14→10mm/ano) resultou de um aumento de ∼33% na massa molar
dos ácidos envolvidos (128→179g/mol) [91].
Além das razões apontadas no início, esses resultados também sugerem a possibilidade
de que a agressividade intrínseca dos vários cortes possam não ser as mesmas, ainda que
os IAT’s envolvidos possam ser similares. Mais especificamente, é possível esperar que
o ácido retido nas frações mais leves da destilação atmosférica (diesel leve, querosene e
nafta), possam vir a apresentar uma maior agressividade do que aquele retido nas
frações mais pesadas da destilação a vácuo (gasóleos). [92]
5.2 O serviço em ambiente quimicamente fragilizante
Foi afirmado que os aços, de uma forma geral, caracterizam-se por possuir elevada
tenacidade, sendo capazes de impor grande resistência à propagação sub-crítica de

122 trincas ou de suportar solicitações a altas taxas de deformação (impacto). Em
conexão, um ambiente ″quimicamente″ fragilizante é aquele que, ao interagir com o
aço, é capaz de não apenas promover a nucleação, mas também o crescimento sub-
crítico de trincas, ou ainda, acarretar reduções na capacidade de suportar impactos.
Dada a sistemática presença do H2S em refinarias nos mais diversos processos, bem
como seu poder de fragilizar o aço via sua hidrogenação, esse tópico faz referência,
inicialmente, aos mecanismos de fragilização, segue com considerações relativas à
interação do aço com o H2S-aq. (ou seja, com o ácido sulfídrico), e de como essa
hidrogenação deve se processar. Finaliza abordando seqüencialmente: as características
apresentadas pelos ″aços avançados ou resistentes ao HIC, isto é, resistentes à
″fissuração induzida pelo hidrogênio″ e ao ″trincamento por corrosão sob tensão em
presença de sulfetos″, ou SSC.
Inicialmente, considerando o aço já hidrogenado, foi proposto (Zapffe) que a
recombinação do átomo de hidrogênio e a formação do gás H2 se dá em cavidades ou
em posições da microestrutura onde o átomo tende a ser retido irreversivelmente. A
formação do gás geraria elevadas pressões internas PH(i)
2 naquelas posições, PH
(i)
2 >>
1atm, produzindo-se um tensionamento interno que seria capaz de promover a
deformação e, por fim, o trincamento do metal. Nestes níveis de pressão, o gás passa a
comportar-se não idealmente. Assim, estritamente, dever-se-ia fazer referência à
fugacidades e não a pressões. Atingido o equilíbrio, o gás hidrogênio H2 acumulado
numa cavidade e o hidrogênio solubilizado intersticialmente, Hi, ½ H2 = Hi, relacionam-
se entre si através da lei de Sievert, , independendo do PH2
externo. Figura 5.8. [48, 93, 94].
O empolamento é resultado do contato com H2S-aq e da recombinação de átomos de
hidrogênio que se difundem nos interstícios da rede cristalina do ferro (com
difusividade de ∼10−5cm2/s), acumulando-se em defeitos de rede e em interfaces
existentes na microestrutura do metal. Como pode ser notado, uma fração do Had torna-
se absorvido, Had → Hab, e outra sofre descarga, 2Had → H2

123
Figura 5.8: Representação esquemática da formação de um ″empolamento″ no ferro..
[93]
Um outro modelo (Troiano) diz respeito ao transporte por difusão do hidrogênio que se
estabeleceria por influência de gradientes de tensões existentes na rede cristalina do aço
entre posições adjacentes. Uma das posições encontrando-se submetida a um estado tri-
axial de tensões (a distensão da rede proporcionada por esse carregamento aumentaria a
solubilidade ao hidrogênio nestas posições), e, a outra, não. É o que ocorre em posições
próximas à frentes de trincas, estas tipicamente solicitadas em estado tri-axial de tensão.
O hidrogênio migraria para aquelas posições e lá, ao invés de se recombinar, interagiria
com o ferro reduzindo as forças de coesão Fe−Fe que resultam de uma ligação metálica,
fragilizariam a região e proporcionariam o crescimento sub-crítico da trinca, como
mostrado na Figura 5.9 [48].

124 Figura 5.9: Considerando a presença prévia do gás H2 numa cavidade produzida por
uma trinca (Zapffe), são mostradas as etapas que antecederiam a
fragilização das ligações Fe-Fe da região submetida a um estado tri-axial
de tensões. São elas: 1. difusão em meio gasoso do H2 até a superfície
″livre″, 2. adsorção, 3. dissociação, 4. absorção e, 5. difusão no sólido.
[48]
Além de possibilitar um crescimento sub-crítico de trincas, o hidrogênio reduz a
tenacidade à fratura do aço (KIC). Simbolicamente, KIC (aço não hidrogenado) → KICH
(aço hidrogenado), sendo KIC > KICH. Assim, na medida em que o carregamento nas
proximidades de uma trinca embebida de tamanho 2a, com fator de forma c, submetida
à solicitação σ, é dado por, K = c. . .aI σ π , e como uma propagação instável é prevista
ocorrer sempre que KI ≥ KIC ou KI ≥ KICH, tal falha será de ocorrência mais provável em
um aço hidrogenado. Considerações similares serão feitas a seguir ao se abordar a
nucleação de fissuras HIC.
Um terceiro modelo (Petch) sugere que a presença do hidrogênio adsorvido em
superfícies livres, tal como as associadas à fissuras internas, favoreceria
termodinamicamente uma propagação sub-crítica uma vez que, nestes casos, a criação
de novas superfícies seria energeticamente favorecida. Por fim, um quarto modelo
emprega como argumento o aumento da mobilidade de discordâncias (Beachem) para
explicar fragilizações. Mediante esse modelo, em presença do hidrogênio, mesmo
carregamentos de baixa magnitude seriam suficientes para acarretar intenso movimento
de discordâncias no aço, e a fragilização seria decorrente da intensificação da
deformação plástica localizada [48]. O aumento da mobilidade das discordâncias
decorreria da redução da barreira de energia elástica existente entre discordâncias
vizinhas devido a formação de ″atmosferas de Cottrell″, resultado direto da interação
discordância−hidrogênio. [95]
Ocorre com freqüência em refinarias o contato do aço carbono com meio aquoso
alcalinizado pela amônia. São as águas residuais ou "águas ácidas″, na qual se
desenvolve:
NH3 + H2O → NH4OH → NH4+ + OH− (→ pH∼8/9), (2.45)

125
contendo ainda os ânions HS− e S2− (devido as dissociações, H2S → HS− + H+ e
HS−→S2−+H+), e CN− (HCN → CN− + H+), todos eles oriundos de ácidos bastante
fracos.
Da interação aço×água residual, ou mesmo aço×filmes aquosos condensados em um
ambiente (vapor) contaminado com H2S, NH3, decorre a corrosão, a adsorção e a
absorção, pelo aço, de uma fração do hidrogênio catódico adsorvido em sua superfície.
Para ser mais específico, ao corroer-se, o ferro gera os elétrons necessários à redução do
sulfeto-ácido, ânion resultante da primeira dissociação do ácido sulfídrico, que é
favorecida num ambiente alcalino. Dessa forma, numa primeira etapa, tem-se H2S → H+
+ HS−, o que é seguido por, Fe → Fe2+ +2e−, em paralelo com HS− + e− → Had + S2−, ao
qual pode seguir-se, Fe2+ + S2− → FeS, composto insolúvel em meio alcalino, que, sob
certas circunstâncias, pode apresentar características protetoras. Além da redução do
HS−, é possível ainda a redução direta do H2S, particularmente em meios pobres em
amônia ou não alcalinos, ambientes em que o ácido sulfídrico não deve se dissociar de
forma importante por efeito do íon comum. Nestes casos, poder-se-ia ter:
H2S+e−→Had+HS−, seguindo-se, H2S+Had+e−→H2(a)+HS− (mecanismo de descarga de
Volmer-Heyrovsky), ou H2S+e−→Had+HS−, seguindo-se 2Had→H2(a) (mecanismo de
descarga de Volmer-Tafel) [93, 96].
Entretanto, em qualquer dos casos, gera-se intermediariamente um hidrogênio que numa
primeira instância, é adsorvido nas áreas catódicas. Na presença do HS−, e de outros
inibidores da reação de despolarização, 2 Had → H2, a absorção do hidrogênio adsorvido
pelo metal, Had → Hab, passa a ocorrer [96]. Nestes ambientes, atribuí-se ao
abaixamento da energia de ligação Fe-Had o aumento da energia de ativação associada à
reação de descarga. [97]
O conjunto de condições que determinam uma maior ou menor agressividade do
eletrólito relativamente à sua capacidade hidrogenante (pH, temperatura, concentração
ou pressão parcial de H2S, natureza e a concentração de contaminantes), podem ser
resumidas na forma de uma “atividade superficial do hidrogênio”, aH. Assim, se o aH for
alto, alto deverá ser o grau de recobrimento do hidrogênio θH = cH/(cH)∞, em que cH
indica a concentração do hidrogênio adsorvido Had relativamente a (cH)∞, a
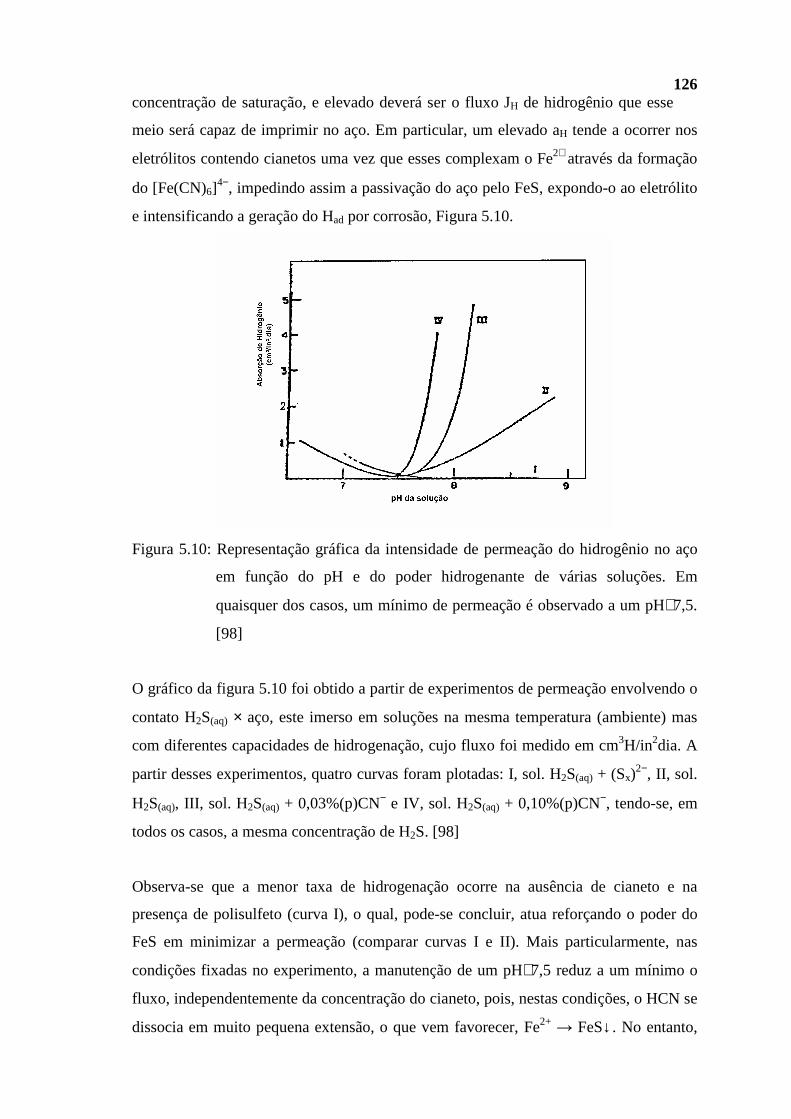
126 concentração de saturação, e elevado deverá ser o fluxo JH de hidrogênio que esse
meio será capaz de imprimir no aço. Em particular, um elevado aH tende a ocorrer nos
eletrólitos contendo cianetos uma vez que esses complexam o Fe2+ através da formação
do [Fe(CN)6]4−, impedindo assim a passivação do aço pelo FeS, expondo-o ao eletrólito
e intensificando a geração do Had por corrosão, Figura 5.10.
Figura 5.10: Representação gráfica da intensidade de permeação do hidrogênio no aço
em função do pH e do poder hidrogenante de várias soluções. Em
quaisquer dos casos, um mínimo de permeação é observado a um pH∼7,5.
[98]
O gráfico da figura 5.10 foi obtido a partir de experimentos de permeação envolvendo o
contato H2S(aq) × aço, este imerso em soluções na mesma temperatura (ambiente) mas
com diferentes capacidades de hidrogenação, cujo fluxo foi medido em cm3H/in2dia. A
partir desses experimentos, quatro curvas foram plotadas: I, sol. H2S(aq) + (Sx)2−, II, sol.
H2S(aq), III, sol. H2S(aq) + 0,03%(p)CN− e IV, sol. H2S(aq) + 0,10%(p)CN−, tendo-se, em
todos os casos, a mesma concentração de H2S. [98]
Observa-se que a menor taxa de hidrogenação ocorre na ausência de cianeto e na
presença de polisulfeto (curva I), o qual, pode-se concluir, atua reforçando o poder do
FeS em minimizar a permeação (comparar curvas I e II). Mais particularmente, nas
condições fixadas no experimento, a manutenção de um pH∼7,5 reduz a um mínimo o
fluxo, independentemente da concentração do cianeto, pois, nestas condições, o HCN se
dissocia em muito pequena extensão, o que vem favorecer, Fe2+ → FeS↓. No entanto,
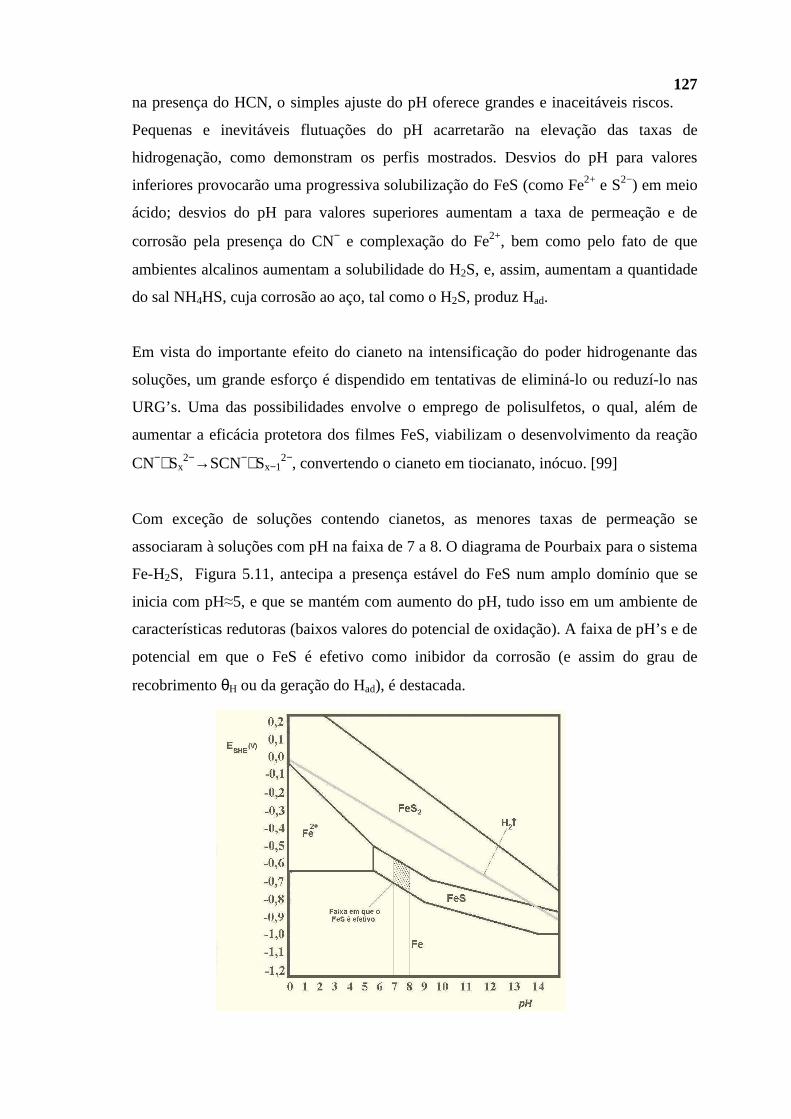
127 na presença do HCN, o simples ajuste do pH oferece grandes e inaceitáveis riscos.
Pequenas e inevitáveis flutuações do pH acarretarão na elevação das taxas de
hidrogenação, como demonstram os perfis mostrados. Desvios do pH para valores
inferiores provocarão uma progressiva solubilização do FeS (como Fe2+ e S2−) em meio
ácido; desvios do pH para valores superiores aumentam a taxa de permeação e de
corrosão pela presença do CN− e complexação do Fe2+, bem como pelo fato de que
ambientes alcalinos aumentam a solubilidade do H2S, e, assim, aumentam a quantidade
do sal NH4HS, cuja corrosão ao aço, tal como o H2S, produz Had.
Em vista do importante efeito do cianeto na intensificação do poder hidrogenante das
soluções, um grande esforço é dispendido em tentativas de eliminá-lo ou reduzí-lo nas
URG’s. Uma das possibilidades envolve o emprego de polisulfetos, o qual, além de
aumentar a eficácia protetora dos filmes FeS, viabilizam o desenvolvimento da reação
CN−+Sx2−→SCN−+Sx−1
2−, convertendo o cianeto em tiocianato, inócuo. [99]
Com exceção de soluções contendo cianetos, as menores taxas de permeação se
associaram à soluções com pH na faixa de 7 a 8. O diagrama de Pourbaix para o sistema
Fe-H2S, Figura 5.11, antecipa a presença estável do FeS num amplo domínio que se
inicia com pH≈5, e que se mantém com aumento do pH, tudo isso em um ambiente de
características redutoras (baixos valores do potencial de oxidação). A faixa de pH’s e de
potencial em que o FeS é efetivo como inibidor da corrosão (e assim do grau de
recobrimento θH ou da geração do Had), é destacada.

128 Figura 5.11: Diagrama de Pourbaix para o sistema Fe-H2S-H2O, 25ºC; ausência de
cianetos e de polisulfetos. [adapt., 11].
Em princípio, todas essas condições encontram-se naturalmente presentes nas águas
geradas em uma refinaria; é isenta tanto de oxigênio como de outros compostos com
poder oxidante relevante. Por outro lado, tal constatação evidencia uma das limitações
do diagrama de Pourbaix. Embora haja previsão termodinâmica do recobrimento do aço
pelo sulfeto em pH’s>5, não é possível fazer previsões relativas à sua eficácia.
Devido à microestrutura típica dos aços laminados, o hidrogênio absorvido
intersticialmente, ao difundir-se, pode ser retido irreversivelmente acumulando-se em
interfaces como são as existentes entre a matriz metálica e as inclusões alongadas de
sulfeto de manganês, essas orientadas “paralelamente” à superfície da chapa ou à
direção de laminação. Posições como essas viabilizam reações do tipo Hab + Hab → H2
(teoria do tipo Zapffe), e, neste caso, o gás resultante pressurizará e promoverá um
carregamento mecânico que tende a “descolar” a inclusão da matriz metálica, dando
origem a uma cavidade na posição e tensionando, simultaneamente, as proximidades,
estas fragilizadas pelo hidrogênio (teoria do tipo Troiano). Simplificadamente, esse é o
mecanismo de nucleação de ″trincas induzidas pelo hidrogênio″ (HIC).
Efetivamente, um HIC só será nucleado se for atingido um valor de pressão na cavidade
PH(i)
2, o qual guarda correspondência direta com a concentração de equilíbrio do
hidrogênio solubilizado intersticialmente, cHi, que acarrete um carregamento KI que seja
maior ou igual a um carregamento crítico para a nucleação de HIC’s, KIHIC, sendo,
K =2
.P . .aI H(i)
2ππ , (2.46)
a expressão derivada do carregamento no modo I que se estabelece nas extremidades de
uma trinca de comprimento 2a quando pressurizada a PH(i)
2, e ( )K =
2. P . .aIHIC H
(i)
Thc2π
π ,
em que ( )PH(i)
Th2 desígna a pressão a partir da qual um HIC é nucleado e 2ac é o
comprimento crítico associado a uma dada inclusão [100]. A seguir, na Figura 5.12, dá-
se uma representação esquemática de uma trinca de comprimento 2a.

129
Figura 5.12. Representação esquemática de uma cavidade de tamanho 2a pressurizada a
pH2 em equilíbrio com o hidrogênio dissolvido intersticialmente cH. Tais
condições acarretam um carregamento KI em sua extremidade. [100]
É interessante notar que parece haver uma correspondência direta entre, por exemplo, a
concentração de cianetos e o ″nível″ do PH(i)
2 que pode potencialmente desenvolver-se
numa dada interface. Poderia ser afirmado que o aumento da concentração de cianetos
acarreta o aumento da atividade superficial do hidrogênio, ou, ↑%CN− ⇒ ↑aH ⇒ ↑JH ⇒
↑cHi ⇒ ↑ PH(i)
2, tornando mais provável ter-se PH
(i)
2 ≥ ( )PH
(i)
Th2. [101]
Na medida em que as inclusões distribuem-se a vários níveis relativamente à espessura
da chapa, o campo de tensões de uma pode interagir e se superpor ao de outra vizinha, o
que, juntamente com pressurizações subseqüentes, pode terminar por interligá-las via
ruptura da matriz, matriz que deve se apresentar fragilizada pelo hidrogênio, isto é, com
a tenacidade reduzida relativamente ao mesmo aço não hidrogenado. A repetição desse
processo leva ao ″trincamento em degrau″ (SWC), essencialmente paralelo à superfície
da chapa [102], o qual pode ou não evoluir para um “empolamento” (HB). Figuras 5.13
a e b. Na Figura 5.13a, a disposição da fissura decorre da orientação aproximadamente
paralela à superfície livre das interfaces retentoras de hidrogênio existentes na
microestrtura dos produtos siderúrgicos obtidos por método termo-mecânico, laminação
no caso. Sua posição é mais próxima da superfície que em serviço fazia contato com o
ácido. Na Figura 5.13b, o perfil T operou imerso em um ambiente encontrado no topo
de uma torre extratora de H2S/NH3 de uma unidade de águas residuais. Nota-se que as
regiões deformadas já se apresentam fissuradas, resultado da fragilização imposta pelo
hidrogênio.

130
(a) (b)
Figura 5.13: (a) Aspecto de uma trinca em degrau (SWC) nucleada no casco em aço A-
285GrC de um vaso de pressão cuja parede (1/2″) manteve contato com
o ácido sulfídrico em serviço. (Regap, MG) e, (b) Perfil T em aço AISI
410 apresentando múltiplos empolamentos (HB) resultado do contato
com condensado contendo proporções desconhecidas de H2S/NH3, além
de (possivelmente), HCN. (Regap, MG)
Enquanto um HB tende a ocorrer a partir dos SWC’s gerados em chapas finas, ou a
partir daqueles próximos à superfícies livres de chapas grossas, uma situação de maior
gravidade (por tornar mais provável a ocorrência de vazamentos de um gás letal),
existirá se o HIC nuclear em regiões particularmente tensionadas, caracteristicamente
num nível superior ao que decorre de um simples carregamento pela pressão interna em
vasos de pressão. Neste caso, ao invés de gerar-se um SWC, a interligação entre HIC’s
tenderá a ocorrer perpendicularmente à superfície da chapa, dando origem a um
″trincamento induzido pelo hidrogênio e orientado por tensão″, SOHIC, Figura 5.14. Na
Figura 5.14, as fissuras se orientam perpendicularmente à superfície da chapa e podem,
por exemplo, provir da interação das bordas de um empolamento (HB) ou de uma trinca
em degrau (SWC) com o campo de tensões residuais associados com ZTA’s de soldas.

131
Figura 5.14: Disposição e morfologia típicas de uma trinca induzida pelo hidrogênio e
orientada por tensão (SOHIC). [103]
Os aços carbono, ″resistentes ao HIC″, devem ser produzidos com base em práticas
específicas. São aços cujas amostras devem ser testadas quanto à resistência que
oferecem à nucleação do HIC, essa expressa por índices relacionados com a extensão
longitudinal do trincamento nucleado num corpo de prova prismático, quando o mesmo
é exposto a um ambiente hidrogenante, como o índice CLR (″crack length ratio″), e ao
longo da espessura, e o índice CTR (″crack thickness ratio″) [103]. Exemplificando,
para um serviço ″classe A″, o aço deve apresentar um CLR≤5% e CTR≤1,5% além
%S≤0,003, %P≤0,010, entre várias outras exigências [104]. Um serviço classe A é
aquele com elevado poder hidrogenante, usualmente contendo cianetos (>20ppm), além
do H2S (>50ppm) e água livre.
Uma empresa projetista de unidades de hidrotratamento seleciona o aço carbono com
base em ambientes existentes em operação normal e não usual. Será requerido um aço
″H2S resistent″, aço classe C da N-1706, se, em operação, houver água livre com H2S <
50ppm ou ″traços″ de H2S em fase vapor, ou ainda, se há água livre fora de operação
com H2S nela dissolvido. Será requerido um aço ″HIC resistent″, próximo ao aço classe
A da N-1706, se se produzir água livre em operação normal com H2S > 50ppm ou se
houver quantidades significativas de H2S em fase vapor, em qualquer dos casos
independentemente da presença de meio orgânico. [104]

132 Devem ser utilizados métodos de fabricação de aço que incorporem a normalização
e a desoxidação de modo a se produzir um aço totalmente homogeneizado e acalmado,
que modifiquem a morfologia das inclusões através da adição de elementos
globulizantes, que promovam a redução dos teores de enxofre e fósforo com a
conseqüente redução da fração volumétrica de inclusões, bem como o ajuste da
composição química do aço, de modo a ter-se um CE limitado a 0,43 (existe a
necessidade de se compatibilizar a resistência mecânica do aço após o TTAT com a
espessura e o CE). Enquanto que as alterações microestruturais induzidas por tais
práticas deve aumentar a resistência do material ao HIC, SWC e HB, ela reduz, em
contrapartida, a resistência ao SOHIC, [105]. O controle do CE, juntamente com a
aplicação do TTAT, são indicados para minimizar a possibilidade de ter-se
microestruturas indesejáveis e, assim, durezas elevadas a nível da ZTA, o mesmo se
passando com componentes submetidos a deformação a frio. Reduz-se assim,
fortemente, a possibilidade da nucleação de trincas por SSC, bem como o trincamento
por SOHIC, mecanismo de trincamento também dependente dos níveis de
tensionamento existentes em serviço. Figuras 5.15 a e b. Na Figura 5.15a, pode-se notar
um forte bandeamento ou alinhamento da microestrutura, esta composta por uma matriz
ferrítica e perlita (escura), em cuja interface o H tende a ficar aprisionado. Na Figura
5.13b, o aço foi submetido a um tratamento termo-mecânico (não especificado) que
elimina por completo o alinhamento ferrita-perlita; adicionalmente tem as inclusões
globulizadas mediante adição de Ca. Interessante notar que como o bandeamento
microestrutural deixa praticamente de existir, parece favorecer-se a interconexão dos
HIC’s ao longo da espessura, eventualmente dando origem a SOHIC’s se existirem
entalhes ou se o nível de carregamento mecânico for suficiente para tal.
(a) (b)

133 Figura 5.15: (a) Microestrutura de uma chapa 1/2″ de aço A516Gr70 normalizado-
convencional, CE=0,42/%C=0,22-%S=0,020-%P=0,027. (Aumento:
200×) e, (b) Microestrutura ferrito-perlítica de uma chapa 1/2″ aço
A516Gr70 normalizado-resistente ao HIC, CE=0,37/%C=0,15-
%S=0,001-%P=0,005. (Aumento: 200×). [105]
Por fim, será feita menção à interação do hidrogênio com estruturas de ″baixa
temperatura de transformação″ (o exemplo clássico é a martensita), como são aquelas
susceptíveis de ocorrência nas ZTA’s de cordões de solda dos aços carbono e aços liga
principalmente. Dessa interação e em conformidade com um dos mecanismos
discutidos, pode resultar a nucleação e, neste caso, a propagação de fissuras por
corrosão sob tensão por sulfetos, SSC, denominação que se justifica pelo fato de que um
mecanismo de dissolução anódica gerador de Had se faz presente. Em particular, um
trincamento SSC foi o responsável pela explosão de uma torre absorvedora de H2S de
uma corrente de propano, fato que ocasionou 17 fatalidades e prejuízos superiores a 100
milhões de dólares. [106]
No que diz respeito às ações mitigadoras, a NACE 0175, [107], recomenda que a dureza
em juntas soldadas se mantenha limitada em 22HRC. Para o aço carbono, essa condição
poderia ser conseguida facilmente mediante a aplicação de um TTAT a 600ºC, que,
além de revenir a região, proporcionaria também, o alívio de tensões. Entretanto,
segundo a própria NACE, em lugar do tratamento térmico pode-se aplicar
procedimentos de soldagem que sejam capazes de assegurar os níveis de dureza
recomendados pois, neste mecanismo, diferentemente dos mecanismos de trincamento
por corrosão sob tensão tradicionais, a interação do hidrogênio com microestruturas
pouco dúcteis, e não tensões residuais, apresentam papel relevante. [108]

134 CAPÍTULO 6
6.1 Corrosão nas destilações atmosférica e vácuo
6.1.1 Sistemas de topo. Corrosão em baixa temperatura
Dada à sempre importante presença de sais inorgânicos hidrolizáveis no petróleo,
fundamentalmente, MgCl2/CaCl2/NaCl, em proporção próxima daquela encontrada na
água do mar [11], juntamente com a forma hidratada dos dois primeiros,
MgCl2.6(H2O)/CaCl2.2(H2O) [109, 110], mais sedimentos e água ("água de formação"
existente nos poros da rocha-reservatório), o controle da corrosividade em meio aquoso
nas unidades de destilação atmosférica e vácuo, mais especificamente, nos seus
condensadores, na tubulação e nos vasos de seus “sistemas de topo”, todos com
metalurgia a base do aço carbono, tem início com a dessalgação do petróleo. Para tanto,
o petróleo após ser pré-aquecido até cerca de 150°C numa ″bateria″ de trocadores de
calor, recebe uma injeção adicional de água pela simples razão de que os sais e demais
contaminantes inorgânicos tendem a se solubilizar nessa fase.
A remoção desses sais se constituí, de fato, na principal dentre todas as outras ações
tomadas para mitigar não apenas os processos corrosivos em baixa temperatura, mas
também para evitar obstruções em trocadores de calor, a formação de coque e no
interior dos tubos de fornos de processo (pois sais catalisam a formação do coque), e
ainda, evitar a desativação de catalisadores (UFCC).
Nos vasos dessalgadores, dado o tempo de residência necessário, a água (salmoura) por
ser mais densa do que o óleo, dele se separa, processo que tem início com o
coalescimento de gotículas inicialmente emulsionadas e distribuídas homogeneamente
no petróleo. O fenômeno da desimulsificação, apesar de favorecido
termodinamicamente pois se passa com a redução da energia interfacial água-óleo, é
assistido pela aplicação de intensos campos elétricos, os quais ao polarizar as gotículas
de água, induzem o seu coalescimento por atração eletrostática [8]. Entretanto, por
razões diversas, a eficiência desse processo é apenas limitada, ou seja, uma fração do sal
e água inicialmente contidos no petróleo deixam de ser retidos nas dessalgadoras (a
dessalgação é feita em duplo estágio na Regap). Pode-se citar como exemplo a
dificuldade em se manter uma interface perfeitamente definida entre o óleo e a salmoura

135 de modo a se evitar arrastes, Figura 6.1. Pode ser notado na Figura 6.1 a tubulação
que interliga o topo das torres atmosférica e vácuo com os seus respectivos ″sistemas de
topo″. Nota-se, ainda, num primeiro plano, os dois vasos dessalgadores (trata-se de uma
dessalgação feita em dois estágios), e ao lado uma bateria de trocadores de calor pré-
aquecedores do petróleo a ser dessalgado.
Figura 6.1: Vista panorâmica das torres atmosférica e vácuo, essa última mais ao fundo
e com maior diâmetro. (Regap, MG)
Segue-se que, nos níveis de temperatura existentes no petróleo a jusante das
dessalgadoras, em particular após uma segunda bateria de pré-aquecimento e no
ambiente do forno, isto é, estando o petróleo aquecido entre cerca de 200 e 400°C, as
reações de hidrólise dos sais remanescentes à dessalgação têm início .
São elas:
MgCl2 + 2 H2O = Mg(OH)2 + 2 HCl, (4.1)
CaCl2 + 2 H2O = Ca(OH)2 + 2 HCl, (4.2)
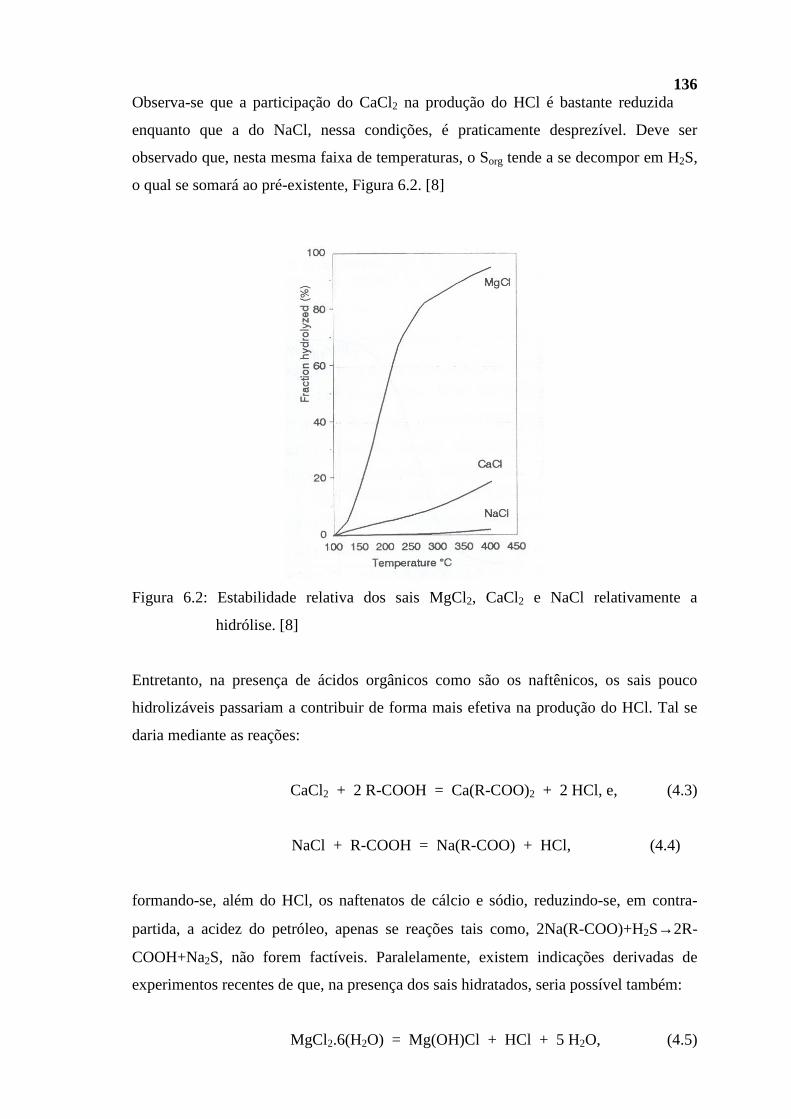
136 Observa-se que a participação do CaCl2 na produção do HCl é bastante reduzida
enquanto que a do NaCl, nessa condições, é praticamente desprezível. Deve ser
observado que, nesta mesma faixa de temperaturas, o Sorg tende a se decompor em H2S,
o qual se somará ao pré-existente, Figura 6.2. [8]
Figura 6.2: Estabilidade relativa dos sais MgCl2, CaCl2 e NaCl relativamente a
hidrólise. [8]
Entretanto, na presença de ácidos orgânicos como são os naftênicos, os sais pouco
hidrolizáveis passariam a contribuir de forma mais efetiva na produção do HCl. Tal se
daria mediante as reações:
CaCl2 + 2 R-COOH = Ca(R-COO)2 + 2 HCl, e, (4.3)
NaCl + R-COOH = Na(R-COO) + HCl, (4.4)
formando-se, além do HCl, os naftenatos de cálcio e sódio, reduzindo-se, em contra-
partida, a acidez do petróleo, apenas se reações tais como, 2Na(R-COO)+H2S→2R-
COOH+Na2S, não forem factíveis. Paralelamente, existem indicações derivadas de
experimentos recentes de que, na presença dos sais hidratados, seria possível também:
MgCl2.6(H2O) = Mg(OH)Cl + HCl + 5 H2O, (4.5)

137
o mesmo se dando com o CaCl2.2(OH),
CaCl2.2(H2O) + H2O = Ca(OH)Cl + HCl + 2 H2O, (4.6)
formando-se o hidroxi-cloreto de magnésio e de cálcio, respectivamente. Tais
compostos são estáveis até cerca de 380°C, podendo decompor-se a partir daí conforme,
Mg(OH)Cl = HCl + MgO, (4.7)
o mesmo se dando com o Ca(OH)Cl [109]. Como temperaturas superiores a 380°C são
encontradas na destilação a vácuo, é possível que tais reações sejam as principais
responsáveis, juntamente com a hidrólise do CaCl2 residual, pelo HCl lá formado.
Tendo-se em conta a agressividade do HCl-aq ou ácido clorídrico ao aço carbono (ácido
formado a partir da dissolução do HCl no condensado formado no topo das destilações),
é usual injetar-se a jusante das dessalgadoras e a montante da 2º bateria de pré-
aquecimento, compostos ″inibidores de hidrólise″. Uma base forte tal como o NaOH é
freqüentemente empregada. Com essa injeção objetiva-se ter a seguinte reação:
MgCl2/CaCl2 + 2 NaOH = 2 NaCl + Mg-Ca(OH)2, (4.8)
convertendo-se o sal hidrolizável em um virtualmente não hidrolizável. Entretanto,
existem efeitos colaterais que limitam a quantidade da soda que pode ser injetada (até
∼5lbsoda/103bbl≅14ppm) [8, 11]. Por exemplo, o sódio presente no RV, carga das
unidades de coqueamento retardado, tende a catalisar a formação de coque nos tubos
dos fornos daquela unidade, reduzindo fortemente o ″fator de utilização″ daquela planta
dada a necessidade de se promover o descoqueamento dos tubos numa freqüência mais
elevada do que a que seria necessária na sua ausência. Há também a possibilidade da
ocorrência da fragilização/corrosão cáustica se, por qualquer razão, a hidroxila
acumular-se de modo a ter-se localmente um pH>12. Por fim, há o aumento da
quantidade de um cloreto não-volátil e não-hidrolizável o que torna possível ″exportá-
lo″ via resíduos para outras plantas, incluindo a própria destilação a vácuo. Assim
sendo, discute-se hoje a conveniência do emprego desse composto. [111]

138
Mesmo com o uso da soda no seu limite, a experiência demonstra que não se consegue
uma completa neutralização do sal remanescente. Segue-se ser necessário complementar
o ″ataque″ ao HCl-aq no sistema de topo e no próprio ambiente da torre. O costado em
aço carbono da torre, topo-cilindro e calota, é revestido internamente com tiras de
monel, Cu-30Ni, liga resistente à corrosão promovida pelo ácido. Evidentemente, a
neutralização dos efeitos do HCl-aq será muito mais importante se nenhum inibidor de
hidrólise pós-dessalgadora for empregado.
Em virtude de sua volatilidade, o HCl (Peb ≅ −85°C), e também o H2S (Peb ≅ −61°C),
juntamente com compostos orgânicos cuja massa molar varia entre aquela
correspondente à do diesel até o GC se separam na zona de vaporização, fundo da torre
atmosférica, local onde se produz um resíduo líquido contendo ou podendo conter, entre
outros não voláteis, os compostos NaCl, Mg(OH)Cl e Ca(OH)Cl. Esse "resíduo
atmosférico" (RAT), faz nível no fundo e é mantido em ebulição a cerca de 370°C,
devendo seguir como carga para a destilação a vácuo. A fração vaporizada constituída
por hidrocarbonetos, C1 até ∼C20 mais HCl, H2S e vapor d’água, seguem em fluxo
ascendente até o topo da torre, ∼110°C.
Dependendo do ponto de ebulição, o hidrocarboneto poderá ou não ficar retido numa
bandeja numa dada elevação da torre. Então, o mesmo deverá ocorrer com o HCl e o
H2S. O hidrocarboneto, o sulfeto e o cloreto ficarão retidos, e deverão seguir como
cortes laterais, se a temperatura do líquido lá acumulado for inferior ao de ebulição do
hidrocarboneto. Ainda assim, é possível que as frações diesel, querosene e nafta,
retenham e terminem por arrastar uma pequena fração de hidrocarbonetos mais leves,
incluindo-se aí o HCl e o H2S.
Portanto, antes de seguir como produto, esses compostos mais leves devem ser
removidos dessas frações e serem re-enviados à torre. Isso é feito injetando-se vapor
d′água em torres retificadoras laterais de forma a reduzir-se a pressão parcial dos
componentes vaporizados lá existentes, obtendo-se dessa forma, um efeito equivalente
ao de reduzir-se a pressão total. Tal redução irá favorecer a vaporização dos mais leves,
os quais, juntamente com o vapor d′água, serão reconduzidos à torre [5]. O mesmo deve
então ocorrer com o HCl e com o H2S.

139
Esse conjunto de características (alta volatilidade e a retificação) constituem-se na razão
pela qual tanto o cloreto como o sulfeto tendem a concentrar-se no topo da torre
atmosférica, e seguir como efluente “de topo”. Isso ocorre em paralelo com
hidrocarbonetos leves, C1 a ∼C8 (esses mais pesados arrastados) e vapor d’água oriundo,
principalmente, do vapor de retificação, o qual pode iniciar a condensação mesmo antes
de alcançar os condensadores de topo, isto é, ainda na tubulação, Figura 6.3.
Figura 6.3: Vista do sistema de topo (vaso acumulador e trocadores-condensadores
instalados em plataformas) de uma destilação atmosférica. (Regap, MG)
Uma vez definido o aço carbono como material construtivo do sistema de topo, o
procedimento recomendado para manter o processo corrosivo generalizado em níveis
aceitáveis (≤5mpy), processo ocasionado pelo contato do aço com o H+ e Cl−
(resultantes da dissociação do HCl) e HS− (resultante da primeira dissociação do H2S),
fundamenta-se em criar condições nas quais o aço possa passivar-se pela formação do
filme ...HSFeSH, o que se daria segundo as reações:
Fe2+ + HS− → (FeSH)+, e (FeSH)+ + HS− → (HSFeSH), (4.9)

140 o qual pode polimerizar-se na superfície do aço [11]. A formação, a estabilidade e a
eficácia protetora do filme será dependente da pressão parcial de H2S, da concentração
de cloretos e do pH. Contudo, sendo o condensado ácido e rico em cloretos e sulfetos, o
aço não deveria passivar-se pois, neste caso, forma-se um complexo solúvel,
...HSFeSH...+2H++2Cl−→Fe(H2S)22+(Cl−)2. Em meio neutro, o cloreto competiria com o
sulfeto-ácido na formação do filme, tornando-o muito pouco efetivo. Há, ainda, a
possibilidade do ataque direto do ácido ao aço, Fe + 2HCl → FeCl2 + 2 H, e, na
presença do H2S, ter-se, FeCl2 + H2S → 2 HCl + FeS. O ácido pode também ser
regenerado por hidrólise do próprio produto de corrosão, FeCl2 + H2O → 2 HCl + FeO.
É, então, necessário reduzir a concentração do cloreto, buscando uma dessalgação mais
eficiente, muito particularmente necessária no caso da não injeção de soda, neutralizar o
ácido, e ainda, injetar compostos inibidores (tipicamente aminas fílmicas) que possam
reforçar a capacidade protetora do sulfeto. [8, 11]
Tradicionalmente, a neutralização do HCl é realizada através da injeção de soluções
aquosas contendo bases nitrogenadas, tal como a amônia (NH3 + HCl → NH4+ + Cl−),
ou como as aminas (R-NH2 + HCl → R-NH3+ + Cl−). Como decorrência da
neutralização com a amônia, sais de amônio higroscópicos, NH4+Cl−, são gerados em
fase vapor podendo ocasionar ″corrosão sob depósito″, se precipitados. [11, 110].
Em qualquer dos casos, o neutralizante deve ser dosado para que se promova a
formação de uma solução tampão com pH na faixa 6,0±0,5. A presença de soluções
desta natureza (constituída por uma base fraca e por seu sal), é necessária ao considerar-
se a possibilidade de que as concentrações do HCl e do H2S possam vir a apresentar
importante variabilidade. A faixa de pH’s selecionada tem por objetivo minimizar a
formação de depósitos a base de FeS nos condensadores, uma vez que essa é a
condição na qual o sulfeto ainda se solubiliza na solução. [112]
Para a seleção do neutralizante, as seguintes considerações são feitas. [110]
- ter uma baixa tendência à formação de sal. A uma dada temperatura, essa tendência
pode ser avaliada pelo valor da constante de estabilidade, k, considerando-se que a
precipitação do sal deverá ocorrer apenas se k < P PHCl n× , onde PHCl e Pn é a
pressão parcial do HCl e do neutralizante, respectivamente;

141
- possuir um ponto de ebulição relativamente baixo. Essa característica deve garantir
volatilidade suficiente de modo que o vapor do neutralizante possa atingir todos os
pontos onde o cloreto gasoso se fizer presente. Por exemplo, a amônia entra em
ebulição a −33ºC, enquanto a morfolina (C4H9ON) o faz apenas ao atingir 129ºC;
- força da base. A base deve ser fraca de forma a dar origem a soluções tampão. Por
exemplo, a amônia (pKb ≅ 4,7 a 25ºC), é mais fraca do que a di-amina de etileno,
EDA, (pKb ≅ 3,3 a 25°C) e mais forte do que a morfolina (pKb ≅ 5,7 a 25°C);
- constante de equilíbrio vapor-líquido, (VLE). Essa constante indica de que forma o
neutralizante deve se distribuir nas fases líquida e vapor (razão vapor/água).
Considerando que o HCl possui um baixo VLE, as primeiras gotas a se formar
deverão dar origem a eletrólitos muito agressivos devido a um baixo pH. Assim, é
de interesse que o neutralizante também possua um baixo VLE. Exemplificando, o
VLE da amônia é 346 enquanto que o da morfolina é de ∼15 e o da EDA é de ∼1.
Logo, esses dois últimos compostos neutralizarão com muito maior eficácia as
primeiras gotas a se formar.
Além da neutralização, é usual e bastante importante compor o esquema de combate à
corrosão no sistema de topo com a injeção de água de lavagem. A injeção deve ser feita
a montante dos condensadores e teria como função diluir e aumentar o pH do
condensado, solubilizar sais eventualmente precipitados (os sais de amônio são todos
eles muito solúveis), e seqüestrar ainda em fase vapor os constituintes ácidos, reduzindo
suas pressões parciais e a possibilidade de precipitação.
Para ilustração, na Figura 6.4, mostra-se o estado de um feixe em aço carbono (A-214)
de um condensador pertecente ao sistema de topo de uma unidade de destilação
atmosférica, tal como se apresentava para inspeção/manutenção após operar por cerca
de 7,8 anos. Dado o avançado estado de deterioração do feixe, o mesmo veio a ser
substituído.

142
Figura 6.4: Condição física do feixe (A-214) de um dos condensadores do topo de uma
destilação atmosférica. Fluido interno aos tubos: água de refrigeração,
fluido do casco ou externo aos tubos: hidrocarbonetos+ HCl+H2S+vapor,
efluente do topo. (Regap, MG)
O resíduo proveniente do fundo da torre atmosférica, o RAT, que corresponde a ∼50%
da vazão da carga da unidade considerando o perfil de petróleos tipicamente
processados na Regap, é direcionado ao forno da unidade de destilação a vácuo. Tendo
sua temperatura elevada até cerca de 400°C, acessa, via linha de transferência, a zona de
vaporização da torre dessa unidade, a torre-vácuo. Lá, sob pressões da ordem de
30mmHg, tem-se a vaporização de seus constituintes voláteis, os quais seguem em
fluxo ascendente, em direção ao topo da torre. Restará um resíduo líquido composto por
asfaltenos e hidrocarbonetos com elevada massa molar que é mantido no ponto de
bolha. É o resíduo de vácuo, RV, que deve seguir como carga da planta de coqueamento
retardado (corresponde a ∼25% da vazão do RAT). O hidrocarboneto vaporizado, a
depender de seu ponto de ebulição, será liqüefeito e coletado em panelas, obtendo-se
duas retiradas laterais. Na destilação a vácuo, tais cortes são genericamente
denominados gasóleos e um deles deve seguir como carga para o craqueamento
catalítico (gasóleo pesado ou GOP), e o outro será incorporado ao ″pool″ de diesel
(gasóleo leve ou GOL), devendo seguir para o hidrotratamento.
Diferentemente do que ocorre na torre atmosférica, no vácuo não há retificação lateral
nem retirada de produto de topo; obtém-se, apenas, uma pequena quantidade de
hidrocarbonetos leves que será separada da água condensada posteriormente. Esse

143 ambiente se caracteriza por possuir uma baixa pressão total, e portanto, uma baixa
pressão parcial de H2S, fato que deve tornar mais difícil a passivação do aço com
sulfetos. Em contrapartida, baixa deverá ser também a pressão do HCl, possivelmente
gerado através da interação NaCl/CaCl2 + R-COOH, bem como por hidrólise dos sais
Mg(OH)Cl-Ca(OH)Cl, o que, a princípio, torna possível o controle da corrosividade
injetando-se apenas neutralizantes. Entretanto, deficiências nesta injeção podem levar a
falhas por corrosão ácida em intervalos extremamente curtos, Figura 6.5.
Figura 6.5: Trecho de feixe (A-214) severamente corroído de um condensador de topo
de uma unidade de destilação a vácuo tal como se apresentava após operar
por apenas cerca de 1 ano. (Regap, MG)
6.1.2 Sistemas de fundo. Corrosão em alta temperatura (~200 – ∼∼∼∼400°C)
Os equipamentos de ambas as destilações, nos trechos submetidos à temperaturas entre
cerca de 200 e 400ºC, encontram-se propensos a sofrerem corrosão por sulfetação e por
ácidos naftênicos, ressaltando-se a necessidade e a importância de se conhecer a forma
pela qual os IAT′s se distribuem nos cortes.
Parece haver consenso de que a forma mais efetiva de se combater ou de impor
resistência a ambos os processos corrosivos se faz pela via ″metalúrgica″. Aços de alta
liga ao cromo (AISI 410s, por exemplo) são indicados para resistir à sulfetação ao passo
que os inoxidáveis austeníticos ligados ao molibdênio (AISI 316, por exemplo) são
indicados para o serviço com petróleo ou cortes ácidos, aço que, em paralelo, apresenta
também uma excelente resistência à sulfetação [8].

144 Não obstante, a injeção de inibidores da corrosão naftênica tem sido considerada e
testada. Pode-se citar como exemplo, os ésteres fosfóricos que ao reagirem com o
Fe2+/Fe3+ dão origem a filmes protetores cujas características decorrem da estabilidade
das ligações Fe-P [86].
Tendo presente que os óleos atualmente extraídos de diferentes e importantes campos
produtores no Brasil têm associado um relativamente baixo teor de enxofre, <1%, com
um importante nível de acidez, >0,5mgKOH/g, este com tendência crescente, a
importância da corrosão naftênica em refinarias pode ser evidenciada observando-se a
montagem dada a seguir na Figura 6-6 a-b-c-d-e. Trata-se do retrato da situação
encontrada em uma torre a vácuo e na tubulação que a interliga ao forno após uma
campanha de apenas 5 anos processando-se um RAT com IAT ∼1,2mgKOH/g, este
oriundo de uma carga composta por petróleos nacionais com ∼0,8mgKOH/g.
Procurou-se exemplos de casos em que o ataque pelo ácido tivesse sido claramente
assistido por efeito mecânico, Figuras 6.6 b-c-e, bem como outros em que inverso
tivesse ocorrido; ou seja, com componente mecânica nula, Figura 6.6 a-d. Em particular,
a Figura 6.6.a-d apresenta o dano sofrido por recheio existente na torre na elevação
GOR (AISI 316 com %Mo<2,5) e por sua estrutura de suportação (AISI 304),
respectivamente. Em nenhum dos casos se observou a existência de fatores mecânicos
na ocorrência da degradação. Trata-se de um típico ataque associado à condensação de
gotículas de ácido. A especificação de materiais com Mo alto é necessária (AISI 317 ou
mesmo o AISI 904, este com 4,5%Mo nominal), muito especialmente para os elementos
de recheio, Figura 6.6 a, dada as espessuras tipicamente encontradas nesses
componentes, usualmente da ordem de décimos de milímetro [85].

145
Figura 6.6: Esquema ilustrativo de uma torre e linha de transferência forno – torre de
uma unidade de destilação a vácuo, e as diferentes formas de ataque pelo
ácido naftênico.

146 Na Figura 6.6.b, fica evidente que o movimento giratório da válvula induzido pelo
fluxo ascendente de gases, acelerou significativamente o desgaste da bandeja no ponto
de contato, tendo sido ambos, bandeja e válvula, fabricados em aço AISI 316. Embora o
teor de molibdênio da bandeja tenha se revelado inferior a 2,5%, não se acredita que
neste caso este tenha se constituído num fator preponderante. Portanto, sendo viável do
ponto de vista do processo, deve-se adotar bandejas providas de borbulhadores fixos
(principalmente) juntamente com a especificação apropriada da metalurgia, AISI 316
com %Mo>2,5.
A Figura 6.6.c evidencia a ocorrência de ataque naftênico assistido por impingimento
ocasionado por gotas de hidrocarboneto bruscamente vaporizado. O ataque ocorreu em
um aço AISI 410s (revestimento do costado), na imediata adjacência da calha de entrada
de carga. Embora a metalurgia do revestimento se mostre claramente inadequada, tiras
previamente instaladas em aço AISI 316 na região (teor de molibdênio desconhecido),
tem apresentado resistência a essas condições.
Na Figura 6.6.e, é mostrada a ocorrência de corrosão-erosão severa localizada nas
adjacências de um cordão de solda em posição ″sobre-cabeça″, trecho reto de tubulação
horizontal (linha de transferência) em aço 5Cr-0,5Mo com 46″ (Figura 2.85). Além de
uma metalurgia inadequada, atribuí-se a ocorrência do desgaste à formação de
turbulência localizada. Uma raiz saliente de solda teria provocado perturbações no fluxo
propiciando a formação de uma condição localizada de elevada erosividade. Deve-se,
então, procurar eliminar quaisquer protuberâncias internas e, principalmente, evitar o
contato direto do fluido com aços do tipo Cr-Mo nestas posições.
Na Figura 6.7, é apresentado o fluxograma simplificado de uma unidade de destilação
com capacidade de processamento de 12.000 m3/dia (∼75.000 barris/dia), onde são
identificados a metalurgia e os locais de ocorrência de diversos tipos de degradação do
aço.

147
6.1.3 Fluxograma (destilação atmosférica e vácuo)
Figura 6.7: Fluxograma simplificado de processo de uma unidade de destilação
(atmosférica e vácuo).

148 CAPÍTULO 7
7.1 Corrosão em unidades de craqueamento catalítico
7.1.1 Área quente (reação, regeneração e fracionamento)
Buscando, principalmente, a produção de GLP e de nafta a partir de uma carga com
baixo valor comercial, o gasóleo (GOP/vácuo-coque) pré-aquecido a cerca de 300°C,
faz contato com catalisador finamente pulverizado (Al 2O3-SiO2) a cerca de 650ºC. Esse
contato ocorre na base de uma tubulação vertical de ″subida″ (″riser″), com cerca de 1m
de diâmetro e ∼40m de altura, local em que o gasóleo, ao ser bruscamente vaporizado,
leva à ″fluidização″ da mistura. Ambos seguem em fluxo ascendente devido ao
diferencial de pressão existente entre os equipamentos que compõe a planta;
essencialmente o próprio riser, um vaso separador ou reator, um vaso regenerador e dois
″tubos de descida″ de catalisador (reator→regenerador e regenerador→riser). Tal
contato acarreta, ao longo do riser, a clivagem das ligações C-C e C-H das moléculas
constituintes da carga. É, assim, produzida uma complexa mistura de hidrocarbonetos
com massa molar em geral mais leve do que as que compunham a carga original
incluindo hidrocarbonetos insaturados, os quais devem seguir para separação por faixa
de ebulição em uma torre fracionadora. Inevitavelmente, gera-se também um resíduo
sólido com baixa razão H/C (coque), que tende a se depositar sobre as partículas de
catalisador, desativando-o. Por sua vez, os compostos sulfurados, nitrogenados e
clorados contidos na carga devem deixar o reator, juntamente com o hidrocarboneto
craqueado, sob as formas dos compostos H2S, NH3, HCN, HCl, juntamente com CO-
CO2, gases arrastados pela corrente de catalisador regenerado.
Ao final do riser, ou seja, em sua extremidade superior, o hidrocarboneto deve ser
separado do catalisador, o qual deve ser regenerado para utilização subseqüente. Para
tanto, encontra-se montado em sua extremidade superior um reator cujo aumento brusco
de volume propicia a separação inicial da mistura, ao fazer com que haja redução da
velocidade dos vapores em ascensão. A separação é finalizada em um conjunto de
ciclones internos ao próprio reator, nos quais se tem associados um efeito centrífugo e
gravitacional. Assim, vapor do hidrocarboneto craqueado escoa pelo topo do vaso
separador seguindo até o fundo da fracionadora por uma tubulação de transferência, ao

149 passo que o catalisador segue em fluxo descendente pelo tubo de descida para
regeneração e daí para a base do riser, repetindo-se o ciclo. Antes de acessar o tubo de
descida, o catalisador recebe uma ″lavagem″ com vapor de média pressão para
retificação (remoção) do óleo a ele aderido.
A regeneração do catalisador é realizada através da queima do coque. É executada em
um vaso regenerador através da injeção de ar comprimido à ∼200ºC. Produz-se assim,
uma massa gasosa composta pelos gases N2-CO-CO2+catalisador, aquecida a cerca de
750ºC, que sobe em fluxo ascendente ao longo do regenerador, no qual novos ciclones
visam recuperar o catalisador redirecionando-os ao tubo de descida regenerador→riser.
Na seqüência, produz-se a passagem CO→CO2 na fornalha de uma caldeira e a energia
liberada é aproveitada para gerar vapor de alta pressão ou, alternativamente, na ausência
do CO, pode-se empregar diretamente a energia contida no gás CO2 para essa finalidade
(regeneração realizada com combustão total), Figuras 7.1 e 7.2.
Figura 7.1: Representação esquemática de uma planta de craqueamento catalítico fluido
″side-by-side″. [4]

150
Figura 7.2: Vista geral de uma unidade de craqueamento fluido. A estrutura ao fundo na
cor verde (trata-se de uma tinta termo-sensível) é o vaso regenerador, a
central o vaso separador ou reator, e na extrema direita a torre
fracionadora. Projeto ″side-by-side″. (Regap, MG)
A existência de altas temperaturas, ~750°C, em um ambiente contendo os gases CO-
CO2 (CO2>>CO), e o escoamento de material com caracteríticas erosivas (catalisador),
possibilitam a ocorrência de fluência, de reações microestruturais fragilizantes
(grafitização, sigmatização e carbonetação), bem como erosão. Logo, todas essas
possibilidades devem ser levadas em conta no projeto, na seleção e na proteção
(revestimento anti-erosivo) dos materiais que deverão compor o riser, o vaso separador,
o tubo de descida e o regenerador. Evidentemente, os problemas relacionados deverão
também ser levados em conta nas paradas programadas para manutenção e inspeção. A
Figura 7.3 apresenta a perda de parede da "perna" de um dos ciclones de um reator,
ocasionada por erosão após uma campanha de 5 anos. A espessa camada de cor escura
interna à perna, e adjacências, é coque. Nota-se, devido a inexistência de indicações
externas, que o processo erosivo se desenvolve de dentro para fora, tal como era
esperado.

151
Figura 7.3: Perda de parede por erosão da perna de um dos ciclones (A-387Gr11) do
reator. (Regap, MG).
A malha de ancoragem (usualmente em aço inoxidável da série 300) do concreto anti-
erosivo e isolante que reveste a superfície interna das paredes dos ciclones,
normalmente se encontra sujeita a mecanismos de danos específicos tal como os
relacionados à erosão e à carbonetação catastrófica (Figura 2.60). Evidentemente,
ocorrências como essas podem vir a comprometer a fixação do concreto, e neste caso,
expor diretamente o aço ao ambiente erosivo existente naquela posição e devem
responder pela perda de parede referida acima.
Consequentemente, a eficiência do ciclone irá se reduzir o que acarretará aumento da
presença de catalisador na corrente de hidrocarboneto craqueado que segue para o
fracionamento. Exporta-se, assim, os problemas de erosão para outros pontos da planta,
Figura 7.4.

152 Figura 7.4: Superfície interna de um dos ciclones de um vaso regenerador. Região
escura evidenciando perda do concreto anti-erosivo devido a falha da
malha de ancoragem, AISI 304. (Regap, MG)
Então, além do hidrocarboneto, a massa gasosa que segue via linha de transferência para
a torre fracionadora contém catalisador (em maior ou menor quantidade a depender do
estado físico dos ciclones), e, portanto, protegida com concreto anti-erosivo em sua
parte inicial. Os gases CO2, NH3, H2S, HCN e vapor d’água, tendem a se acumular no
topo da torre, o qual recebe uma injeção de água de lavagem de modo a evitar-se a
deposição de sais.
Os equipamentos e a tubulação que compõe o sistema de topo da fracionadora, além de
encontrar-se sujeito ao trincamento por sulfetos (SSC) nas juntas soldadas em aço
carbono não aliviadas, estão também sujeitos ao trincamento por corrosão sob tensão
por carbonato, eletrólito formado por soluções alcalinizadas pela amônia contendo CO2,
fazendo-se necessário ter-se um pH na faixa de 9 a 11, e um potencial de transição
ativo-passivo próximo a −670mV/SCE, Figura 7.5. Na medida em que o trincamento
por carbonato é assistido por tensão, a simples aplicação de um TTAT, deveria ser
suficiente para prevenir essa ocorrência. [113]
Figura 7.5: Trincas não passantes nucleadas por CST/CO32− na superfície interna e nas
proximidades de um cordão de solda (ZTA) não aliviado. Trecho de
tubulação pertencente ao sistema de topo de uma torre fracionadora de uma
unidade de craqueamento fluido. (Regap, MG)

153 A Figura 7.6 apresenta um fluxograma simplificado de uma unidade de
craqueamento catalítico, seção de reação, de regeneração e de fracionamento (área
quente), cuja a capacidade de processamento é de 3.600m3/dia.
154 7.1.2 Fluxograma (UFCC - área quente)
Figura 7.6: Fluxograma de processo simplificado de uma unidade de craqueamento
catalítico. (Regap, MG)

155 7.1.3 Área fria (recuperação de gases)
Como já se referiu, a mistura de hidrocarboneto craqueado, mais vapor, H2S, HCN,
NH3, CO2, e catalisador arrastado, a uma temperatura próxima a 550°C, deixa o reator
dirigindo-se ao fundo da torre fracionadora. Nesta torre, o fracionamento produz como
corte lateral os óleos de reciclo leve (LCO) e pesado (HCO), este não mostrado na
Figura 5.6, e nafta pesada (NP). Ambos os óleos encontram-se à temperaturas
superiores a 250°C e podem conter algum H2S e catalisador, razão pela qual é possível
ter-se corrosão por sulfetação, corrosão-erosão ou mesmo erosão causadas por esse
fluido. Pelo fundo, escoa um resíduo contendo uma significativa quantidade de
catalisador, que é totalmente reciclado ao ser direcionado à base do riser. É comum
observar-se erosão nos internos das bombas deste sistema. Pelo topo da torre, a cerca de
150ºC, escoam as frações orgânicas leves (C1 a ∼C12), juntamente com contaminantes
inorgânicos já mencionados.
Após resfriar-se na bateria de condensadores do sistema, o efluente do topo, agora
parcialmente condensado, é enviado a um vaso acumulador onde co-existirão três fases:
uma ″fase-gás″ constituída por hidrocarbonetos C1 a C4 e contaminantes, uma fase
orgânica líquida (nafta mais GLP dissolvido denominada ″nafta não estabilizada″),
relativamente pobre em contaminantes, e uma fase aquosa proveniente das injeções de
vapor feita no ″stripper″ do reator e de água de lavagem feita no topo da fracionadora. A
fase aquosa é rica nos íons HS− (oriundos da primeira dissociação do H2S,
H2S→H++HS−), HCO3− (oriundos da primeira dissociação do H2CO3,
H2CO3→H++HCO3−) e CN− (oriundos da dissociação do HCN, HCN→H++CN−), todos
eles originados da dissociação de ácidos fracos sendo, portanto, as favorecidas em meio
alcalino produzido pela hidrólise da amônia, NH3+H2O→NH4++OH−. A Figura 7.7
apresenta uma foto da torre fracionadora da REGAP, e de seu sistema de topo.

156
Figura 7.7: Foto ilustrativa de uma torre fracionadora e do seu sistema de topo. Em
destaque a tubulação que interliga o topo da torre à bateria de
condensadores e daí ao vaso acumulador. (Regap, MG)
Em contato com um eletrólito dessa espécie, o aço tende a ser corroído, originando íons
Fe2+ e elétrons. Como ocorre em soluções contendo íons de várias naturezas e, entre
eles, o HS−, uma possível semi-reação catódica poderia envolver, por exemplo, a
redução do sulfeto ácido, HS−+e−→Had+S2−. Então, a depender dos potenciais de
oxidação, da mobilidade e da concentração dos íons envolvidos, uma ou outra semi-
reação poderá prevalecer.
Considerando que a redução do sulfeto seja a prevalecente, ter-se-ia como produto um
hidrogênio Had adsorvido na superfície metálica podendo, subseqüentemente, ser
absorvido, Had→Hab, em lugar de evoluir como um gás, Had+Had→H2.
Além de tornar mais difícil a despolarização do aço, o íon sulfeto pode também reagir
com o Fe2+ dando FeS insolúvel (em meio alcalino), que, ao precipitar, pode recobrir a
superfície exposta ao eletrólito, reduzindo a intensidade do processo corrosivo, e por
conseqüência, a taxa de permeação do hidrogênio no aço. Assim, todos os fatores
ligados com a estabilidade do FeS devem cumprir um papel na taxa de permeação.

157 Dentre eles, pode-se citar o cianeto. O cianeto age no sentido do aumento da taxa de
permeação por complexar o Fe2+ mediante formação do complexo ferrocianeto
[Fe(CN)6]4−, impedindo ou dificultando, portanto, a passivação do aço. Soluções
aquosas ou filmes condensados contendo mais do que 20ppm de cianetos são
considerados suficientemente ricos neste ânion. As conseqüências da presença do
hidrogênio na rede do aço ferrítico são variadas e sempre deletérias, sendo apresentados
na Figura 7.8. A fragilização do aço se traduz por queda na tenacidade a fratura; o
trincamento pode ser causado por SSC (fissuração por corrosão sob tensão em presença
de sulfetos), sendo para isso necessário a presença de uma microestrutura com dureza
elevada tal como pode ocorrer na ZTA de soldas não aliviadas ou revenidas. O HIC
(fissuração induzida pelo hidrogênio), é o precursor do SWC (fissura em degrau), o qual
pode dar origem ao HB (empolamento), sendo ambas as ocorrências dependentes da
existência de uma microestrura contendo interfaces retentoras de hidrogênio (bandeada
ou alinhada e/ou contendo inclusões do tipo MnS). É também, de certa forma, o
precursor do SOHIC (fissura induzida pelo hidrogênio e orientada por tensão), que além
de uma microestrutura susceptível depende também da existência de carregamentos
mecânicos (residual ou decorrentes de concentradores de tensão) que sejam
suficientemente elevados.
Figura 7.8: Diagrama onde se mostra os possíveis efeitos ocasionados pelo hidrogênio ao aço ferrítico.
Numa refinaria, os equipamentos e tubulações existentes na unidade de "recuperação de
gases″, cuja metalurgia de referência é o aço carbono, encontram-se particularmente
propensos aos danos induzidos pelo hidrogênio. Essa planta consiste em um grande
número de vasos de pressão, torres e trocadores de calor, cuja finalidade é a de

158 possibilitar a separação do GLP do GC e do GLP da nafta. Para tanto, o vapor
oriundo da parte superior do vaso acumulador, uma mistura composta pelo GC e GLP e
contaminantes (incluindo vapor d′água), é comprimida (∼15kgf/cm2.man) e resfriada,
seguindo para um vaso separador de alta pressão, mostrado na Figura 7.9.
Figura 7.9: Trecho de uma planta de recuperação de gases de uma UFCC. Destaque para
o vaso separador de "alta". (Regap, MG)
Neste vaso, a água e os hidrocarbonetos C3 e C4 apresentam-se liqüefeitos ao passo que
os hidrocarbonetos C1 e C2 ainda mantém-se vaporizados. Estratificam-se, assim, três
camadas: uma aquosa alcalina rica em OH−, NH4+, HS−, CN−, tal como ocorre no vaso
de topo da fracionadora, uma orgânica líquida (GLP) e outra orgânica gasosa (GC),
ambas contendo contaminantes inorgânicos, principalmente essa última. Portanto, todo
o aço subjacente à superfície interna do vaso, mas particularmente aquele em contato
com água e vapor (local onde filmes de água condensada podem ser formados),
encontram-se sujeitos a hidrogenar-se.
A corrente gasosa (C1, C2, contendo C3, C4 e contaminantes) segue para o fundo de uma
torre absorvedora, dita primária, onde será promovido o contato com nafta não
estabilizada efluente do vaso de topo da fracionadora. Nas condições lá existentes, a
nafta deve absorver o hidrocarboneto C3 e C4 presente na corrente. Na seqüência,
devido a um possível arraste de nafta pelo GC, a corrente gasosa é enviada a uma
segunda torre absorvedora (secundária), em que o fluido absorvedor é o LCO, o qual,
após cumprir sua função, retorna à fracionadora, enquanto o GC, livre de
hidrocarbonetos C3 e C4, segue pelo topo para uma planta de tratamento com aminas

159 (DEA) para remoção do H2S (principalmente) e CO2. Tanto a absorvedora primária
quanto a secundária tendem a acumular sulfetos e vapor d′água em seus topos; são
assim produzidos ambientes fortemente hidrogenantes nessas posições.
Por sua vez, a nafta não estabilizada (isto é, nafta+GLP), efluente do fundo da torre
absorvedora primária junta-se com o hidrocarboneto oriundo da descarga do
compressor, é resfriada e segue para o vaso de alta pressão. O efluente líquido desse
vaso, ou seja, nafta + GLP, agora juntamente com algum GC arrastado, segue para duas
torres colocadas em série. A primeira delas, uma torre retificadora, destina-se à
separação da nafta do GC, que retorna ao vaso de alta, empregando-se a energia térmica
do LCO efluente da fracionadora, o que se faz através de ″refervedores″ interligados ao
fundo da torre. Na segunda torre, uma ″debutanizadora″, se processa a separação da
nafta do GLP, empregando-se energia térmica proveniente do HCO, tal como se faz
com o LCO na retificadora. O GLP segue pelo topo para remoção do H2S e dos demais
contaminantes inorgânicos, a semelhança do GC. A nafta, por sua vez, agora isenta de
GLP, segue para remoção de mercaptans na unidade de tratamento Merox. [3, 5].
Uma vez que o H2S, o HCN, o CO2, apresentam elevada tendência de escapar do meio
líquido aquecido, tal como ocorre com os hidrocarbonetos C1 a C4, as correntes de GC e
GLP apresentam-se ricas nestes compostos. Em suma, mais uma vez, verifica-se a
tendência do ″gás ácido″ vir a se acumular nos topos e sistemas de topo de ambas as
torres.
As medidas tomadas para minimizar os danos causados pelo hidrogênio ao aço,
envolvem a injeção de água de lavagem no vaso de topo da fracionadora de modo a
maximizar a remoção dos contaminantes do meio orgânico, a pintura dos vasos, o alívio
de tensões das soldas e, mais recentemente, o emprego dos aços carbono resistentes
[104]. Embora seja menos comum, pode-se injetar uma solução de polisulfetos os quais
reagem com o cianeto formando tiocianato, CN−+Sx2−→SCN−+(x−1)S2−+2e−, evitando-
se assim a despassivação do aço, FeS+6CN−→Fe(CN)6
4−+S2−, e a intensifiicação de sua
fragilização [96].
A Figura 7.10 apresenta o fluxograma simplificado do processo de uma planta de
recuperação de gases, metalurgia e mecanismos de dano, acoplada a uma unidade de
160 craqueamento catalítico. Possui capacidade de processamento de 2.300m3/dia de
nafta, 1.200Nm3/dia de GLP e 8500Nm3/dia de GC.
7.1.4 Fluxograma (UFCC – área fria)
Figura 7.10: Fluxograma simplificado de processo de uma planta de recuperação de
gases de uma unidade de craqueamento. (Regap, MG)

161
7.1.5 Tratamento com aminas (DEA)
Por darem origem a soluções alcalinas pobres condutoras de íons, uma amina pura (uma
base fraca), não é corrosiva ao aço, seja ela uma amina primária, R-NH2, por ex.
monoetanolamina ou MEA, uma amina secundária, R2-NH, por ex. dietanolamina ou
DEA, ou terciária, R3-N, por ex. metildietanolamina ou MDEA, sendo R = HOCH2CH2,
R2 = (HOCH2CH2)2. [114]
Soluções aquosas a base de aminas ou alquil-alcanolaminas são largamente empregadas
como soluções absorventes na remoção do gás ácido e descontaminação do
hidrocarboneto. Das aminas listadas acima, encontra-se frequentemente o emprego de
uma solução aquosa contendo DEA a 20-30%, para o tratamento do GC e do GLP,
efluentes da seção de recuperação de gases, isto é, contendo H2S predominantemente,
HCN, CO2 (e, eventualmente, outros contaminantes).
O processo se fundamenta na reversibilidade da reação exotérmica de neutralização
ácido-base Amina+HÁcido=AminaH++Ácido−, ou, H2S+R2-NH=R2-NH2++HS− e
CO2+2R2-NH=R2-NH++R2-NCOO−+H+, quando particularizadas para a interação DEA
com o H2S e CO2, respectivamente. A reação direta representa a remoção dos
contaminantes do hidrocarboneto (seja ele GC ou GLP), processo que se passa em
torres absorvedoras a baixas temperaturas (∼35°C), locais em que o GC e o GLP se
deslocam em sentido ascendente e em contra-corrente com a solução DEA. Como
resultado, escoa pelo topo um hidrocarboneto descontaminado (o GLP será ainda
enviado para extração de mercaptans), e pelo fundo de ambas as torres escoa uma amina
″rica″ em H2S, que segue para a seção de regeneração. Em antecedência, a solução DEA
rica é aquecida às custas do resfriamento da DEA regenerada ou ″pobre″ (em H2S),
efluente de uma torre regeneradora, o que se dá em um trocador de calor em que os
fluidos são DEA pobre (casco) e DEA rica (tubos). A regeneração, que corresponde à
reação se processando no sentido inverso ao mostrado anteriormente, ocorre a mais altas
temperaturas (∼125°C) e menores pressões. Parte dessa energia é fornecida por vapor
superaquecido de baixa pressão (∼3,5kgf/cm2man, ∼150ºC) em um refervedor, o que se
faz mediante cessão de calor à solução absorvente. Por fim, a solução regenerada é
reintroduzida no sistema pelo fundo da torre regeneradora para repetição do ciclo de

162 absorção, enquanto o gás ácido, rico em H2S, é recolhido pelo topo, seguindo para
um vaso acumulador após ceder calor a água de refrigeração em um condensador. O gás
ácido, composto fundamentalmente por H2S, é enviado para a unidade de ″recuperação
de enxofre″ (URE).
As soluções de aminas puras não são corrosivas. [114] Entretanto, na medida em que é
empregada em contínua recirculação, esta solução tende a acumular uma série de
compostos de diferentes naturezas, os quais podem modificar amplamente suas
características. Podem, assim, ter reduzida sua capacidade de absorção tornando-se
muito corrosivas. Entre tais compostos, pode-se destacar: os ″sais termicamente
estáveis″ (STE), compostos não susceptíveis de regeneração, os ″produtos de
degradação″ da amina, além da presença de particulado sólido em suspensão
promovedor de processos erosivos. O particulado é basicamente constituído por sulfetos
e óxidos, produtos de corrosão desagregados da parede dos equipamentos/tubulação,
que se somam ao particulado existente na água empregada para a formulação da solução
de reposição. [115]
Independentemente do acúmulo desses compostos, a solução pode tornar-se bastante
agressiva quando combinada com o H2S, particularmente quando aquecida à
temperaturas acima da ambiente, haja visto ser o H2S um composto possuidor de
agressividade intrínseca, seja no sentido da fragilização, seja no sentido da corrosão do
aço. Para uma solução 25-30%DEA, a concentração de H2S da DEA rica deveria
limitar-se a 0,40mol(H2S)/mol(DEA), ao passo que na DEA pobre, o teor de H2S deve
ser de 0,05 a 0,07m/m [114]. Essa última condição é encontrada no sistema de fundo da
torre regeneradora, por onde circula uma amina pobre em H2S, aquecida à temperaturas
da ordem de 120°C. Neste caso, um ″excesso de regeneração″, possivelmente,
acarretando concentrações inferiores a 0,05m/m, poderia levar a formação de uma
solução na qual o H2S ainda se faz presente, mas incapaz de passivar eficientemente o
aço através da formação de sulfetos [116], o que parece particularmente grave na
presença de CO2 dada a possibilidade da formação do ácido carbônico em filmes
condensados. Por outro lado, teores superiores de H2S poderiam acarretar corrosão por
impingimento de gás ácido em posições em que o escape abrupto do gás da solução for
favorecido (″acid gas flashing″) [114], mecanismo evidentemente válido também para a
solução rica.

163
Soluções DEA pobre em H2S, contendo também CO2, com pH entre 8 e 11 à
temperaturas acima da ambiente, são também fortes promotoras do trincamento
intergranular das juntas soldadas não aliviadas em aço carbono ("alkaline stress
corrosion crack" ou ASCC). Verifica-se que essa condição existe quando na região
tensionada se estabece um potencial metal-eletrólito desestabilizador de filmes
protetores. Dada a importância do papel das tensões residuais no processo, essa
ocorrência pode ser evitada mediante aplicação do TTAT. [117] A Figura 7.11
apresenta a ocorrência de corrosão preferencial e o trincamento por ASCC em cordão de
solda não aliviado em um acessório (redução) em aço carbono.
Figura 7.11: Redução concêntrica em aço carbono (A-234GrWPB) por onde fluiu DEA
pobre. (Regap, MG).
Nas plantas que tratam correntes de hidrocarbonetos efluentes de áreas frias de UFCC’s,
a carga a tratar, além do H2S, contém também CO2 e pode conter o HCN, entre outros
contaminantes. Neste caso, a agressividade da solução de aminas ao aço tende a
acentuar-se, com o seu uso, progressivamente. Além da possibilidade da formação
direta do ácido, o CO2 proporciona a degradação da DEA. Esse processo inicia com a
formação do ácido carbâmico NCOOH e se encerra com a formação do hidroxi-etil
piperazina (HEP), numa seqüência (irreversível) do tipo: R2-NH+CO2→R2-
NCOO−+H+→HED (hidroxi-etil oxazolidona)→THEED (tri-hidroxi-etil
etilenodiamina)→HEP. [114, 115]
Além do CO2 e do HCN, é também possível a contaminação do sistema com oxigênio.
Diferentemente dos contaminantes anteriores, o O2 é, tipicamente, admitido na planta

164 através da própria solução de reposição, usualmente devido a formas indevidas de
armazenamento que permitiriam o contato com o ar. O oxigênio é particularmente
deletério pois, além contribuir diretamente para o ataque ao aço ao dar suporte às semi-
reações catódicas (O2+2H2O+4e−→4OH−, em meios alcalinos), pode também ocasionar
a formação de sais estáveis, o que teria início com a oxidação da amina. Tal processo
pode resultar na formação dos ácidos oxálico (COOH)2 (pKa∼4,3 a 25°C) e glicólico,
CH2OH.COOH (pKa∼3,9 a 25°C), ambos corrosivos. Além disto, pelo fato de
possuirem uma força muito superior à do H2S (pKa∼7,0 a 25°C) e do CO2 (pKa∼6,4 a
25°C), ao interagir com a DEA, deverão dar origem a amino-sais não susceptíveis de
regeneração [118]. Por sua vez, o HCN atuaria complexando o Fe2+ impedindo a
passivação do aço pelo FeS, ou, poderia reagir com o H2S formando o ácido tiociânico
HSCN (pKa∼0,85 a 25°C), o qual, pelas razões já apontadas, também leva a formação
de sais estáveis. [115] A Figura 7.12 apresenta um processo de corrosão no casco de um
refervedor de uma torre regeneradora de solução DEA. O processo corrosivo pode estar
associado à presença em excesso de sais estáveis e/ou de compostos oriundos da
degradação da solução.
Figura 7.12: Corrosão intensa que incidiu na totalidade da parte superior do casco
cilíndrico (A285-C) de um refervedor de uma torre regeneradora de
solução DEA. (Regap, MG)
Tomando o aço carbono como material de referência (Regap), parece ser do consenso
entre os especialistas que um bom desempenho dessa planta (baixos custos de reposição
da solução juntamente com uma adequada capacidade de absorção e baixa
corrosividade), passa, essencialmente, pela manutenção de uma solução absorvente

165 “livre” (livre no sentido de que não se ultrapasse certos valores de referência), dos
particulados, dos sais estáveis e dos produtos de degradação.
Por essas razões encontram-se disponíveis vários métodos de limpeza dessas soluções,
incluindo dispositivos que possibilitam a remoção contínua dos STE’s em linha [119].
De uma forma geral, é recomendado manter a concentração dos sais estáveis e produtos
de degradação limitados a 2,5% (p). [115]
A Figura 7.13 apresenta o fluxograma simplificado de uma planta de tratamento de GLP
e de GC em solução DEA. Possui capacidade de tratamento de 2.000Nm3/dia de GLP.
São mostrados, também, a metalurgia e os mecanismos de dano tipicamente
encontrados nessas plantas.
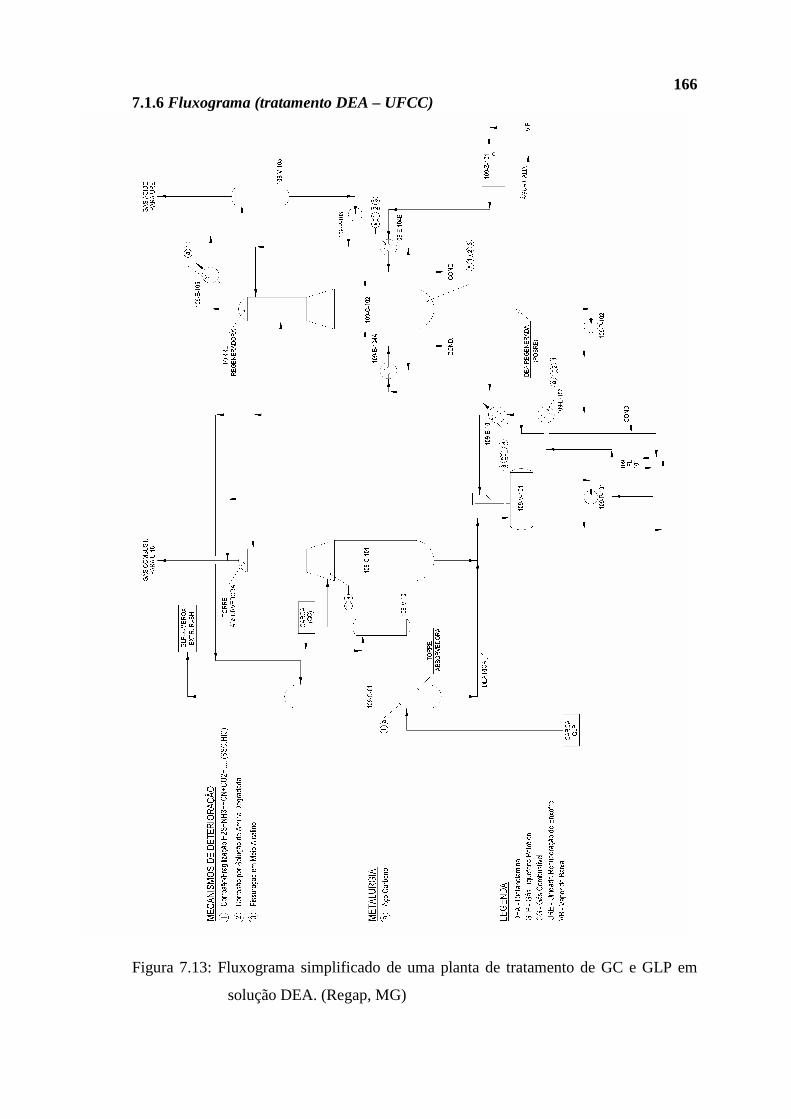
166 7.1.6 Fluxograma (tratamento DEA – UFCC)
Figura 7.13: Fluxograma simplificado de uma planta de tratamento de GC e GLP em
solução DEA. (Regap, MG)

167 CAPÍTULO 8
8.1 Corrosão no hidrotratamento (HDT/HDS)
Em refinarias de petróleo, a saturação dos compostos insaturados e o enquadramento do
nível de impurezas de um dado corte à legislação ambiental, e, ainda, a redução da
corrosividade e do odor, em grande parte, efeitos produzidos por compostos sulfurados
e nitrogenados, é feita em unidades de hidrotratamento (HDT) através da hidrogenação
do hidrocarboneto (diesel, nafta, querosene etc). Como referência, em uma das quatro
unidades de hidrotratamento atualmente existentes na Regap, o óleo diesel contendo
cerca de 4.500ppmS – 1.400ppmN inicialmente, deixa a planta contendo apenas cerca
de 130ppmS (redução de ∼97%) - 170ppmN (redução de ∼88%). Para tanto, consome-
se, em média, 2molH2/molS, gerando-se cerca de 67kJ/molH2 na hidrodessulfurização
do hidrocarboneto, possivelmente a mais importante das reações de hidrogenação. [120]
Esse processo se passa em reatores na presença de catalisadores (NiMo), envolvendo
faixas de temperaturas que vão de ∼300 a ∼500°C e pressões entre ∼5 a ∼35MPa,
quando se inclui plantas de hidrocraqueamento, isto é, o craqueamento catalítico
realizado na presença de hidrogênio, [71], não existente na Regap. Geralmente, quanto
mais pesado ou menor for o °API do corte a ser hidrotratado, mais severos (elevados)
deverão ser os valores de temperatura e pressão da planta e maiores deverão ser as
quantidades de ″sub-produtos″ agressivos gerados. A Figura 8.1 apresenta um par de
reatores de uma planta de hidrotratamento.

168
Figura 8.1: Par de reatores de uma planta de hidrotratamento. (Regap, MG)
A hidrogenação do hidrocarboneto insaturado (alcenos) é realizada visando o aumento
da estabilidade química do corte. Pode ser exemplificado pela reação C4H8
(CH3CH2CH=CH2) + H2 → C4H10 (CH3CH2CH2CH3), quando particularizado para o 1-
buteno (→n-butano). Por sua vez, a hidrogenação dos compostos sulfurados (tiois,
dissulfetos, etc), exemplificada pela reação R-SH + H2 → R-H + H2S(g), leva à
formação do H2S como sub-produto qualquer que seja o composto sulfurado, e a
hidrogenação dos compostos nitrogenados (pirrol, piridina, etc) pela reação, R-NH2 +
H2 → R-H + NH3(g), a qual produz amônia, como sub-produto, também em quaisquer
dos casos. [3, 5]. Além dessas reações, a hidrogenação de compostos oxigenados
eventualmente presentes (fenois, ácidos carboxílicos, álcoois, etc), deve acarretar a
formação de vapor d’água, R-OH + H2 → R-H + H2O(g), e ainda a decomposição dos
ácidos carboxílicos/naftênicos, 2R'-R"-COOH + 2H2 → 2R'-H + 2R"-H + H2O(g) +
CO2(g) + CO(g), ao passo que a hidrogenação dos compostos clorados deve levar à
formação do cloreto de hidrogênio como sub-produto, R-Cl +H2 → R-H + HCl (g).
Com referência às reações de hidrodesoxidação, faz-se notar que, independentemente da
acidez da carga, não deve ser esperado o ataque por ácidos naftênicos nos equipamentos
e tubulações posiciondos a montante do reator; em contrapartida, os equipamentos

169 posicionados a montante estarão potencialmente sujeitas ao ataque quando
submetidos à temperaturas superiores a cerca de 220°C, desde que a carga possua IAT′s
superiores a ∼0,5mgKOH/g.
Como decorrência deste conjunto de reações, forma-se, a jusante dos reatores, uma
corrente de hidrocarbonetos com um teor remanescente de compostos sulfurados
orgânicos muito inferior ao teor de compostos sulfurados do diesel-carga (é o ″diesel
metropolitano″), contendo ainda H2 (∼85mol%H2, admitido em excesso), H2S, NH3,
H2O (vapor) e, eventualmente, HCl, aquecidos a ∼355°C e com pressão total de
∼8,7MPa.abs (89kgf/cm2). Subseqüentemente, essa corrente será progressivamente
resfriada em uma bateria de trocadores interligados em série, cedendo calor para o
diesel-carga até que se atinja a temperatura de cerca de 50°C, quando, então, acessa dois
vasos separadores, de alta e de baixa pressão.
Nestes vasos, separam-se uma fase gasosa rica em H2, que segue para reciclo na própria
planta, e uma fase orgânica líquida (diesel) contendo ainda uma quantidade importante
de H2S/amônia, que segue para separação final em uma torre retificadora. Pelo fundo do
vaso, flui uma solução aquosa rica em H2S e em amônia, que segue para a unidade de
tratamento de águas residuais em planta específica. Já na torre de retificação, escoa pelo
topo um gás úmido rico em H2S/NH3, juntamente com algum hidrocarboneto arrastado
(que deve retornar ao topo da torre como refluxo a partir de um vaso acumulador), e,
pelo fundo, a fração de diesel tratado que segue para armazenamento, após resfriar-se,
cedendo calor para o diesel que segue para retificação.
Diante de tais condições, as formas pelas quais falhas podem se desenvolver em
unidades HDT, são bastante variadas o que, por sua vez, serve de referência para a
seleção do aço a ser empregado nestas plantas.
Em particular, o material construtivo dos reatores (2,25Cr-1Mo/Regap), que são, em
operação, continuamente submetidos à temperaturas superiores a 350°C, encontra-se
sujeito a sofrer perdas de tenacidade por fragilização pelo revenido, e a sofrer o ataque
pelo hidrogênio a altas temperaturas. Esta se constituí em uma das razões da
especificação de um aço Cr-Mo. Entretanto, por não resistir à sulfetação por misturas
H2/H2S, o reator tem sua superfície interna revestida com aço inoxidável da série 300

170 (Figura 2.13), o qual, por sua vez, encontra-se propenso a sofrer o trincamento por
IGSCC, fato que leva à especificação de aços estabilizados (AISI 347 usualmente), de
modo a evitar a sensitização durante a fabricação (ciclos térmicos de soldagem e
tratamentos térmicos).
Paralelamente, devido a presença de correntes potencialmente capazes de dar origem a
sais higroscópicos de amônio, NH4HS e NH4Cl, os vários equipamentos existentes a
jusante dos reatores encontram-se sujeitos a sofrer corrosão e/ou obstrução promovida
por esses sais. Nesta posição, entre cerca de 400 e 250ºC, é usual fazer-se uso do aço
inoxidável da série 300 estabilizado ou de baixo carbono (″L″), do aço Cr-Mo e por fim,
do aço carbono, à medida em que a temperatura da corrente se reduz. Então,
considerando-se a elevada criticidade da planta e o usual emprego do aço inoxidável,
um controle severo da presença de cloretos na carga da unidade deve ser realizado, haja
visto a completa incompatibilidade que há entre este ânion, ou o HCl, e o aço inoxidável
austenítico. O tubo representado na Figura 8.2 e mais 82 tubos do feixe do trocador de
calor (~20% do total de tubos), sofreram perfurações em um período de tempo inferior a
dois anos de operação. A espessura de parede dos tubos era de 2,1mm.
Figura 8.2: Evidência da ocorrência de corrosão por pites observada junto à superfície interna de um tubo 3/4″, A-213Gr304L, do feixe de um trocador de calor de uma HDT que operava com um efluente do reator de uma planta de hidrotratamento que continha cloretos na carga. (Regap, MG)
Assim, com o objetivo de se prevenir a precipitação de sais, injeta-se uma ″água de
lavagem″ a montante dos trocadores nos quais, fixadas as pressões parciais do H2S e
NH3, os níveis de temperatura já favorecem a sublimação do sal. Além do mais, na
eventualidade da precipitação dos sais, a quantidade da água deveria ser suficiente para

171 dissolvê-los, considerando a grande solubilidade que ambos apresentam neste meio,
e acarretar concentrações inferiores a 2% do sal NH4H. Procura-se ajustar a velocidade
de escoamento entre 3-6m/s, de modo a minimizar deposições, ao promover o arraste
mecânico do sal eventualmente precipitado, e evitar-se processos de corrosão-erosão,
considerando-se uma metalurgia a base do aço carbono. [39].
A existência de H2S em grandes quantidades, juntamente com vapor que se condensará,
torna possível também a fragilização do aço por meio de mecanismos como HIC, SSC,
SOHIC, particularmente nos vasos separadores e em suas proximidades, e ainda nos
equipamentos e tubulação do sistema de topo da torre retificadora, todos eles em aço
carbono. Nessas posições, o emprego de aços resistentes ao HIC e a aplicação do TTAT
nas juntas soldadas são altamente recomendáveis (N-1706, 2004). Todas esses
mecanismos (potenciais) de falha são apontados a seguir, na Figura 8.3.
A Figura 8.3 apresenta o fluxograma simplificado de uma planta de tratamento HDT.
Possui capacidade de tratamento de 3.500m3/dia de diesel. São mostrados, também, a
metalurgia e os mecanismos de dano tipicamente encontrados nessas plantas.

172 8.1.1 Fluxograma (HDT)
Figura 8.3: Fluxograma simplificado de processo de uma unidade de hidrotratamento de
diesel. (Regap, MG)

173 8.2 Corrosão na unidade de geração de hidrogênio (UGH)
De forma a alimentar com hidrogênio suas quatro unidades de hidrotratamento, produz-
se na Regap cerca de 15.000Nm3H2/h em duas plantas de geração, todo ele derivado da
reforma a vapor do gás natural (~95%CH4) previamente dessulfurizado, operação que é
facilitada pelo conteúdo muito baixo de compostos sulfurados presentes nesta corrente.
O processo se passa em leito de catalisador de níquel (NiO→Ni, o que se dá em
operação devido a presença de ambiente redutor), o qual é introduzido e adensado por
vibração em uma série de tubos verticais ou ″colunas″ com cerca de 11m de
comprimento e 6″ de diâmetro, todos eles fixados por suportes de mola instalados no
topo da câmara de radiação de um forno dotado de queimadores também no teto. Tanto
o gás natural como o vapor, à pressão de ∼20kgf/cm2.man, são alimentados pela parte
superior dos tubos, já pré-aquecidos a ∼450°C. A Figura 8.4 apresenta um forno de
reforma de gás natural a vapor. São 64 colunas em aço A-608GrHP45/Nb distribuídas
em duas fileiras de 32; 16 queimadores no teto dispostos em duas fileiras de 8. O gás de
processo (H2, CO2, CO e vapor) é recolhido pelo fundo em um tubo coletor também em
aço A-608GrHP45/Nb, o qual é interligado por configuração flexível, liga B-407
(Incoloy 800H®), às colunas.
Figura 8.4: Vista de um forno de reforma de gás natural a vapor. (Regap, MG)

174 Ao longo das colunas, as principais reações serão CH4 + H2O(g) → CO + 3 H2, e,
ainda, CO + H2O(g) = CO2 + H2, sendo a primeira, assistida por catalisadores, a reação
irreversível de reforma. Na medida em que essa reação é fortemente endotérmica, há a
necessidade do suprimento de grande quantidade de energia, tipicamente envolvendo
fluxos de calor entre 45 e 90kW/m2 (∼11 e 22kcal/m2.s), tendo por referência a
superfície interna dos tubos. A absorção de calor gera gases que deixam as colunas (H2,
principalmente, CO2, CO, H2O, CH4), aquecidos à temperaturas entre ∼750-850°C e,
paralelamente, temperaturas de parede entre ∼850-920°C. É, portanto, crítica a
manutenção da atividade do catalisador, considerando-se o risco que há da ocorrência
da falha prematura por fluência caso se desenvolvam pontos sobre-aquecidos na parede
metálica (por conseqüência da perda localizada da atividade do catalisador por,
exemplificando, deposição de coque). [121]
Tais tubos, produzidos por fundição centrífuga, têm a espessura de parede dimensionada
[47] com base na tensão mínima que é capaz de levar à ruptura do tubo por fluência
após, usualmente, 100.000h (∼11,4 anos), à temperatura de projeto, esta normalmente
ajustada ∼28°C (50°F) acima da temperatura máxima de operação [122]. Por sua vez, a
tensão mínima de fluência varia amplamente com a metalurgia. Enquanto uma amostra
de um tubo de aço A-608GrHK40 (0,40C-25Cr-20Ni) apresenta como resultado da
aplicação de ensaios acelerados de fluência, uma tensão mínima à ruptura em 100.000h,
a 1.000°C, de 4,9MPa, um aço A-608GrHP45/Nb (0,43C-25Cr-35Ni-0,8Nb) tem essa
tensão aumentada em 63% (8,0MPa). [123]
Na medida em que os tubos dos dois aços são produzidos por fundição centrífuga,
possuindo, portanto, grãos com morfologia acicular e tamanhos similares, o resultado
acima deve decorrer de diferenças na microestrutura proveniente das diferentes
formulações químicas desses dois materiais. Sendo a resistência a fluência dessa classe
de ligas fortemente dependente da fração volumétrica e da distribuição dos carbonetos
em uma matriz austenítica, a performance superior do HP/Nb relativamente ao HK é
creditada à formação de carbonetos do tipo NbC, de menores dimensões do que os dos
tipos, Cr7C3 e Cr23C6, não dando assim origem a uma rede contínua de carbonetos nos
espaçamentos interdendríticos nos quais precipita, o que serve para reduzir a taxa de
escorregamento do grão bem como o nível da tensão que tende se concentrar na
interface matriz-carboneto. Como resultado, o HP apresenta, também, superior

175 ductilidade a quente e uma menor taxa de nucleação de vazios nessas posições. Por
fim, a matriz do HP é possuidora de maior estabilidade relativamente à precipitação de
intermetálicos fragilizantes (fase sigma), apresentando, também, maior resistência à
carbonetação, ambos os efeitos ocasionados pela elevação do teor de níquel. [123]
Embora o cálculo do API seja conservativo, ele faz uso apenas do carregamento
primário decorrente da pressurização do tubo. No entanto, um tubo de um forno
reformador é também submetido em operação a tensões térmicas, tanto mais
importantes quanto maiores forem a espessura de parede. Tal se dá em virtude do maior
gradiente de temperaturas que deve se estabelecer ao longo das paredes mais espessas.
Assim, tubos fabricados com aços mais resistentes à fluência tendem a ser menos
solicitados por carregamentos secundários pelo simples fato de poderem ser
dimensionados com paredes mais finas. [122]
O sistema que recolhe pelo fundo do forno o gás reformado à cerca de 800°C, é
montado fora da câmara de radiação, portanto, não estando sujeito ao contato direto
com chama. O sistema foi concebido para absorver seus próprios movimentos bem
como parte dos movimentos longitudinais de contração e de dilatação das colunas. É
composto por um tubo coletor em A-608GrHP45/Nb, o qual é interligado às 64 colunas
por 64 tubos extrudados com configuração flexível (curvados), em B-407/Incoloy
800H (0,10C-32Ni-20Cr+Al-Ti); um ″Tê″ e um cone de redução ambos em A-
351CT15C (0,10C-32Ni-20Cr-1Nb), produzidos por fundição estática. Transições
geométricas como são as existentes entre uma coluna e um tubo extrudado podem dar
origem à tensões térmicas, e após um número suficiente de paradas (resfriamento) e
partidas (aquecimento) do equipamento, nuclear trincas por fadiga-térmica, Figura 8.5.

176 Figura 8.5: Representação esquemática do conjunto/metalurgia responsável por
recolher o gás efluente de um forno reformador. [ 108].
Cabe ressaltar que o nióbio, não obstante os importantes efeitos benéficos apontados
anteriormente (A-608GrHP/Nb), parece exercer efeitos deletérios na liga A-351 ao
poder precipitar em serviço, juntamente com o silício, sob a forma do composto
intermetálico ″G″, Ni16(Nb,Cr)6Si7. Além de promover importantes reduções na
ductilidade e tenacidade originais da liga à temperatura ambiente e, portanto, na
soldabilidade, impõe também reduções na resistência à fluência. É, assim, possível ter-
se, em decorrência do serviço, o desenvolvimento de trincas por fluência nesses
acessórios, os quais poderiam ter, simultâneamente, a recuperabilidade por solda
comprometida. É ″teoricamente″ possível resgatar a ductilidade da liga aplicando-se um
tratatamento de solubilização a ∼1.100°C. Entretanto, tal tratamento não restaura a
resistência à fluência. [124, 125]
A resistência à fluência pode também sofrer reduções nos tubos que interligam as
colunas ao tubo coletor (Incoloy 800H®). Essa redução ocorreria nas regiões (curvas)
submetidas ao dobramento a frio, portanto, encruadas. Considerando o papel do
tamanho de grão na vida em fluência (as taxas de deformação por fluência são
proporcionais ao inverso do cubo do tamanho de grão), verifica-se que tais regiões
poderiam experimentar um significativo refino do grão devido a uma eventual
recristalização que se desenvolveria em serviço. Tal fenômeno não deve ocorrer se a
estrutura recristalizada for mantida em temperaturas superiores a aquela requerida para
causar unicamente a recristalização. [126, 127]
Como a mistura gasosa efluente do forno reformador contém quantidades importantes
de vapor, a ela associa-se um ponto de orvalho. Na medida em que essa corrente é
resfriada no processo para permitir a posterior separação do H2 do restante dos gases, há
a possibilidade da formação do ácido carbônico. Faz-se necessário assim, a partir de um
dado ponto, passar a se empregar a metalurgia adequada ao contato com eletrólitos
dessa natureza (por exemplo, AISI 304L). Na ocorrência mostrada na foto abaixo,
Figura 8.6, a metalurgia foi alterada (aço liga Cr-Mo → aço inoxidável série 300)
imediatamente à jusante do trocador quando deveria ter sido feita no próprio
equipamento (no feixe tubular, espelho e carretel de saída). Neste trocador, o gás de

177 processo é admitido pelos tubos a cerca de 400°C; após ceder calor para a água de
caldeira (fluido do casco), deixa o trocador a cerca de 170°C. [128]
Figura 8.6: Aspecto do processo corrosivo ácido sofrido por um dos tubos (A-199GrT5-
3/4″) do feixe de um trocador de calor que em serviço fazia contato, pelo
lado interno, com gás efluente de um forno reformador (H2, CO, CO2 e
vapor). Taxa de corrosão:∼4mm/ano. (Regap, MG)
Por fim, já resfriados à temperaturas próximas da ambiente, a corrente H2+CO2 e
demais contaminantes segue para a separação em ″peneiras moleculares″ em unidades
PSA (″adsorção por variação de pressão″), objetivando a obtenção de gás hidrogênio
com alta pureza (o que pode ser feito também com emprego de soluções absorventes a
base de aminas).
Tais peneiras são constituídas de materiais adsorventes, como carvão ativado e alumina,
retentores do CO2, CH4, que são ″transparentes″ ao H2. São dispostos em vasos de
pressão, local em que formam os leitos de adsorção. Por sua vez, a planta é composta
por seis vasos (Regap), interligados em série entre si. Enquanto que a etapa de adsorção
é realizada em pressão relativamente elevada num desses conjuntos (∼14kgf/cm2.man),
a regeneração ou de-adsorção é feita através da redução de pressão do outro conjunto
de vasos (∼0,4kgf/cm2.man), o que vem caracterizar a existência de esforços cíclicos em
operação potencialmente causadores de fadiga mecânica (ou corrosão-fadiga) nesses
equipamentos.

178 A seguir, a Figura 8.7 apresenta o fluxograma simplificado de uma planta de
geração de hidrogênio por reforma a vapor de gás natural. Possui capacidade geradora
de 8.000Nm3/dia de H2. São mostrados, também, a metalurgia e os mecanismos de dano
tipicamente encontrados nessas plantas.

179 8.2.1 Fluxograma (UGH)
Figura 8.7: Fluxograma simplificado de processo de uma unidade de geração de
hidrogênio e PSA. (Regap, MG)

180 CAPÍTULO 9
9.1 Corrosão no coqueamento retardado (UCR)
A denominação ″coqueamento retardado″ designa um processo de conversão que
envolve a passagem, resíduo (obtido na própria unidade) → material sólido (coque) +
hidrocarboneto líquido (gasóleos e nafta) + hidrocarboneto gasoso (GC e GLP), por via
unicamente térmica, isto é, sem a assistência de catalisadores. Por sua vez, o resíduo
empregado como carga é efluente da destilação a vácuo, o ″resíduo de vácuo″ (RV). Em
particular, o ″coque de petróleo″ encontra sua mais importante aplicação na extração do
alumínio da alumina (para cada 1kgfAl extraído por eletrólise emprega-se cerca de
0,4kgfC). A título de ilustração, uma carga com ºAPI de 6-10 (RV→ºAPI=8-14)
apresentou os seguintes rendimentos: ∼35% de coque, ∼58% de hidrocarboneto líquido
e ∼7% de hidrocarboneto gás. [5, 129]. Trata-se, portanto, de um processo com alta
atratividade econômica. A Figura 9.1 mostra. três torres, estando a torre fracionadora
posicionada mais à esquerda. Ao fundo, pode-se observar a estrutura de suportação e
guia das lanças dos reatores e ao centro a chaminé ″única″ de suas duas fornalhas.
Figura 9.1 Vista geral de uma planta de coqueamento retardado (Regap, MG)
Inicialmente, o RV é pré-aquecido em uma bateria de trocadores de calor a valores
próximos a 250°C, o que é feito a partir de troca térmica com os cortes oriundos de uma
torre fracionadora pertencente à própria planta. Pré-aquecida, a carga acessa o fundo

181 dessa torre juntamente com hidrocarboneto gasoso reciclado dos reatores (∼10% da
carga total e a ∼500°C), local em que a fração leve proveniente do hidrocarboneto
gasoso reciclado mais aquele resultante da vaporização da fração leve contida no
resíduo (em muito pequena proporção neste caso), se separa do restante da carga que é
mantida líquida no fundo da torre. Tal como ocorre em uma torre de uma planta de
destilação, essa corrente de leves segue em sentido ascendente, incluindo-se aí o H2S,
fracionando-se em cortes (gasóleos e naftas) ao longo da torre por um mecanismo que
envolve vaporizações e condensações sucessivas em suas várias bandejas. Em
particular, escoa pelo topo da torre nafta leve mais hidrocarbonetos gasosos (C1 até C4)
contaminada com H2S, principalmente. A nafta é separada do gás no vaso de topo,
seguindo a corrente gasosa para compressão e separação, em uma planta muito similar a
uma outra considerada anteriormente (URG-UFCC), incluindo a de tratamento com
aminas (DEA). Contudo, a agressividade dos eletrólitos aquosos produzidos nessas duas
plantas será significativamente inferior devido a inexistência dos contaminantes: CO2,
HCN e NH3.
Sendo o IAT e a temperatura do RV-carga tipicamente baixos, <0,5mgKOH/g e
∼250ºC, a potencialidade da ocorrência de corrosão naftênica na unidade deverá ser
baixa também. Além de baixa, ela deve se restringir à tubulação e aos trocadores de
calor da bateria de pré-aquecimento da carga posicionados a montante da fracionadora,
às elevações na torre e tubulações correspondentes aos cortes do gasóleo médio e
pesado. A redução do IAT com a diluição proporcionada pelo hidrocarboneto reciclado
(IAT=0, devido a degradação térmica do ácido nos fornos da planta), deve ser
compensada pelo incremento da temperatura.
Os processos de corrosão por sulfetação têm que ser considerados para os equipamentos
e sistema de tubulações montados a montante e a jusante da torre, incluindo-se
evidentemente os internos e o casco da própria torre, neste caso, apenas o fundo e as
posições intermediárias que encontram-se submetidas à temperaturas entre 300 e
∼380°C. Por sua vez, a possibilidade da presença conjunta dos ácidos H2S (←Sorg) e,
eventualmente, HCl (devido a passagem NaCl→HCl nos fornos; a seguir), no topo e
sistema de topo da torre, incluindo os equipamentos da unidade de separação de gases,
deve ser considerada. Neste caso, além dos danos resultantes do contato do aço carbono
com o H2S ″úmido″, poder-se-ia ter:

182
Fe + 2 HCl → FeCl2 + H2, (7.1)
o que pode ser seguido por:
FeCl2 + H2S → FeS + 2HCl, (7.2)
regenerando o ácido, à semelhança do que ocorre nos topos das unidades de destilação
atmosférica e vácuo. A presença do ácido pode ser facilmente inferida mediante análise
de cloretos no condensado que lá se forma; ″bota ″ do vaso acumulador.
Considerando que o RV ainda retenha um certo nível de leves, ainda que muito baixo, e
que ele se separe no fundo da fracionadora, deve-se gerar neste fundo um resíduo ainda
mais rico em asfaltenos do que os anteriores. Nota-se que, para chegar a esse resíduo,
foram necessárias as seguintes passagens: resíduo-atmosférico (RAT)→resíduo-vácuo
(RV)→resíduo-coque, o qual é bombeado para sofrer um aquecimento final em um par
de fornos posicionado a jusante da torre. O aquecimento produzido deve ser suficiente
para vaporizar e dar início às reações de formação de coque, devendo-se alcançar cerca
de 500°C na saída dos passos. As ações operacionais tomadas no sentido de retardar
essas reações (injeção de água de caldeira de modo a promover turbulência), justificam
a denominação da planta. [129]
Subseqüentemente, o produto parcialmente vaporizado deve seguir para alimentar de
forma sincronizada um par de reatores (carregamento de um, descarregamento do
outro), locais em que o coque deve efetivamente se formar, resultado da
complementação das reações de coqueamento. A fração de hidrocarbonetos que não
participa das reações de formação de coque, isto é, o hidrocarboneto que permanece
vaporizado escoa pelo topo dos reatores, e segue para reciclo e fracionamento na torre
fracionadora, tal como aludido anteriormente. A Figura 9.2 apresenta um esquema de
uma unidade de coqueamento retardado com seus principais equipamentos: a torre
fracionadora, o forno e os reatores são mostrados. A carga efluente do forno é
direcionada até a base de um dos dois reatores por uma válvula de 3 vias; pelo topo, o
hidrocarboneto que permanece gasoso é reciclado na fracionadora. O sistema de topo da
torre é composto por um trocador-condensador e um vaso separador. Nesta planta, os
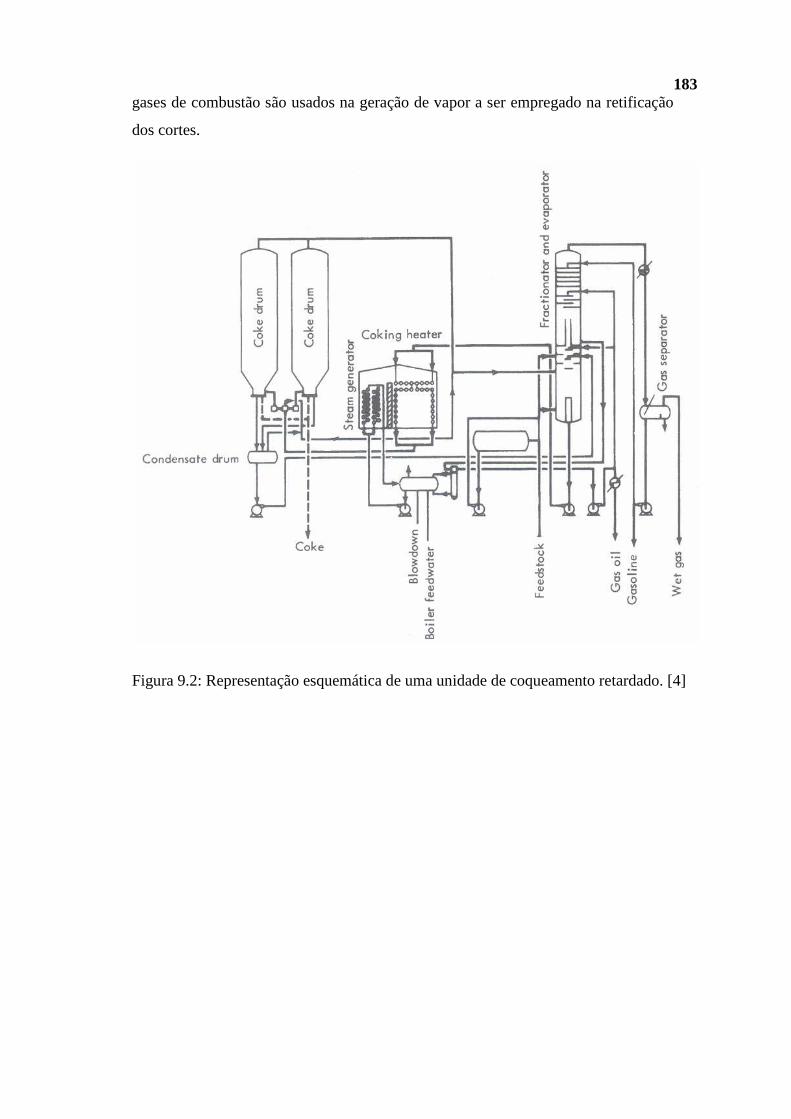
183 gases de combustão são usados na geração de vapor a ser empregado na retificação
dos cortes.
Figura 9.2: Representação esquemática de uma unidade de coqueamento retardado. [4]

184 A Figura 9.3 apresenta uma vista panorâmica do conjunto de reatores de uma
unidade de coqueamento retardado. Para cada par de reatores, há uma válvula de 3 vias.
Figura 9.3: Vista panorâmica do conjunto de reatores de uma unidade de coqueamento
retardado. (Regap, MG)
Além do colapso incremental referido anteriormente, as paredes de um reator
encontram-se sujeitas à sulfetação, razão pela qual são revestidas internamente com aço
inoxidável da série 400. Entretanto, o "fator de utilização" da planta é muito
particularmente impactado pelo seus fornos.
As condições existentes nesses fornos (temperaturas da ordem de 500°C e presença de
vapor d’água), já tornam possível a hidrólise do NaCl, [8],
NaCl + H2O → NaOH + HCl , (7.3)
esse um sal a ser transferido do RV para o resíduo que segue para os fornos. Seriam
duas as razões para que se desse essa transferência: o NaCl entra em ebulição a 1465°C,
ou seja, não deve compor a corrente gasosa que escoa em sentido ascendente na torre-
vácuo; nas condições presentes no fundo dessa torre, ∼430°C, a hidrólise do sal não
deve ocorrer em extensão significativa (ambas as condições se fazem presentes nas
destilações atmosférica e vácuo). Assim, o NaCl deve seguir arrastado pelo resíduo,
carga dos fornos. Então, o HCl gerado nos fornos poderia terminar por acessar os
reatores e, assim, a fracionadora ao ser arrastado pelo hidrocarboneto gasoso a ser
reciclado. Na torre, segue com a corrente gasosa até o topo e depois ao sistema de topo

185 da torre, local onde se concentram os componentes mais voláteis, incluindo o H2S.
Consequentemente, o uso de soda nas destilações com o objetivo de reduzir a
quantidade de sal hidrolizável residual à dessalgação, traz como possível contra-partida
o aumento do NaCl nos resíduos e com isso a possibilidade do aumento da formação de
HCl nas unidades de destilação a vácuo e de coqueamento retardado, sendo que nessa
planta tenderia a se concentrar com a nafta leve (topo). Uma vez que essa corrente
compõe a carga das HDT’s, esse mesmo HCl poderia ser para lá "exportado", gerando
grandes problemas naquela unidade, como a corrosão ocasionada pelo sal NH4Cl.
(Figura 8.2)
O termo coquemento retardado serve também para explicitar a importância do conjunto
de ações no processo destinadas a evitar que haja formação excessiva de coque nos
tubos dos fornos da planta, retardando-o de modo a vir de fato ocorrer nos reatores.
Uma vez que o resíduo é aquecido à temperaturas superiores a de decomposição do
hidrocarboneto, 427°C (800°F), essas ações são: manter o fluido escoando em suas
serpentinas com elevadas velocidades espaciais de modo a reduzir os tempos de
residência do hidrocarboneto nos tubos e a promoção de turbulência via injeção de água
de caldeira. [129]. Ainda assim, a incidência de coque na tubulação da serpentina dessas
fornalhas constituí-se na principal causa da redução do fator operacional, seja pela
necessidade de se proceder descoqueamentos (operação ″idealmente″ levada a efeito em
intervalos regulares ou pela indicação dada por termogramas dessa necessidade durante
a campanha), seja pela ocorrência evidentemente indesejável da ruptura em operação de
um ou mais tubos.
A importância da manutenção de uma serpentina tanto quanto possível ″limpa″, pode
ser ilustrada considerando que o coeficiente de condutividade térmica do coque é da
ordem de 1/50 do coeficiente de condutividade de um aço ferrítico, ou seja, algo em
torno de 1,2W/m.K (1kcal/h.m.ºC). Assim, a presença de coque aderido à superfície
interna dos tubos de uma fornalha traz como conseqüências imediatas o sobre-
aquecimento e o aumento do acúmulo de danos por fluência juntamente com o aumento
das taxas de desgaste da parede do tubo por oxidação externa. Em particular, sendo a
taxa de transferência de calor por unidade de superfície de troca térmica dos tubos na
seção de convecção de um desses fornos de ∼17kW/m2 (∼4kcal/s.m2), o acúmulo de
coque (verificado em ocorrência recente), tendo alcançado ∼15mm de espessura, pode

186 ter acarretado temperaturas próximas a 680°C na posição, significativamente
superior àquela que serve de referência como sendo a máxima alcançável em operação
estando os tubos isentos de coque, 479°C. Por sua vez, sobre-aquecimentos desse nível
já seriam suficientes para reduzir a vida do tubo em regime de fluência para apenas
cerca de 1,5 anos quando se tem por referência a metalurgia lá especificada (tubos em
aço 5Cr-1/2Mo) e um carregamento mecânico resultante da pressurização interna de
13MPa [130]. A Figura 9.4 mostra a falha de um dos tubos da convecção de um forno
de uma unidade de coqueamento retardado.
Figura 9.4: Aspecto externo de um tubo A-335P5/3,5″, pertencente a seção de
convecção de um forno de uma unidade de coqueamento retardado que
veio a falhar em operação devido ao acúmulo de coque internamente.
Ruptura promovida por fluência assistida por oxidação externa. (Regap,
MG)
As Figura 8.7 e 8.8 apresentam o fluxograma simplificado de uma planta de
coqueamento retardado, fracionamento e seção de reação, respectivamente. Possui
capacidade de processamento de 3.800m3RV/dia. São mostrados, também, em ambos os
casos, a metalurgia e os mecanismos de dano tipicamente encontrados nessas plantas.
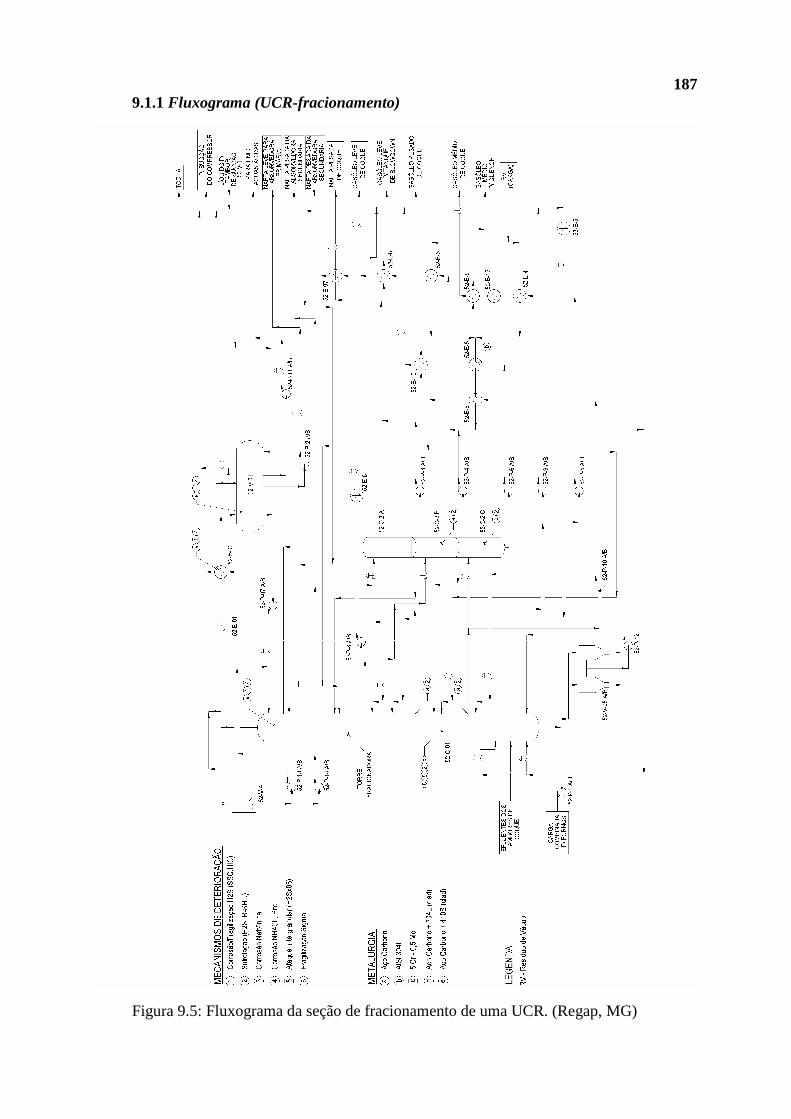
187 9.1.1 Fluxograma (UCR-fracionamento)
Figura 9.5: Fluxograma da seção de fracionamento de uma UCR. (Regap, MG)
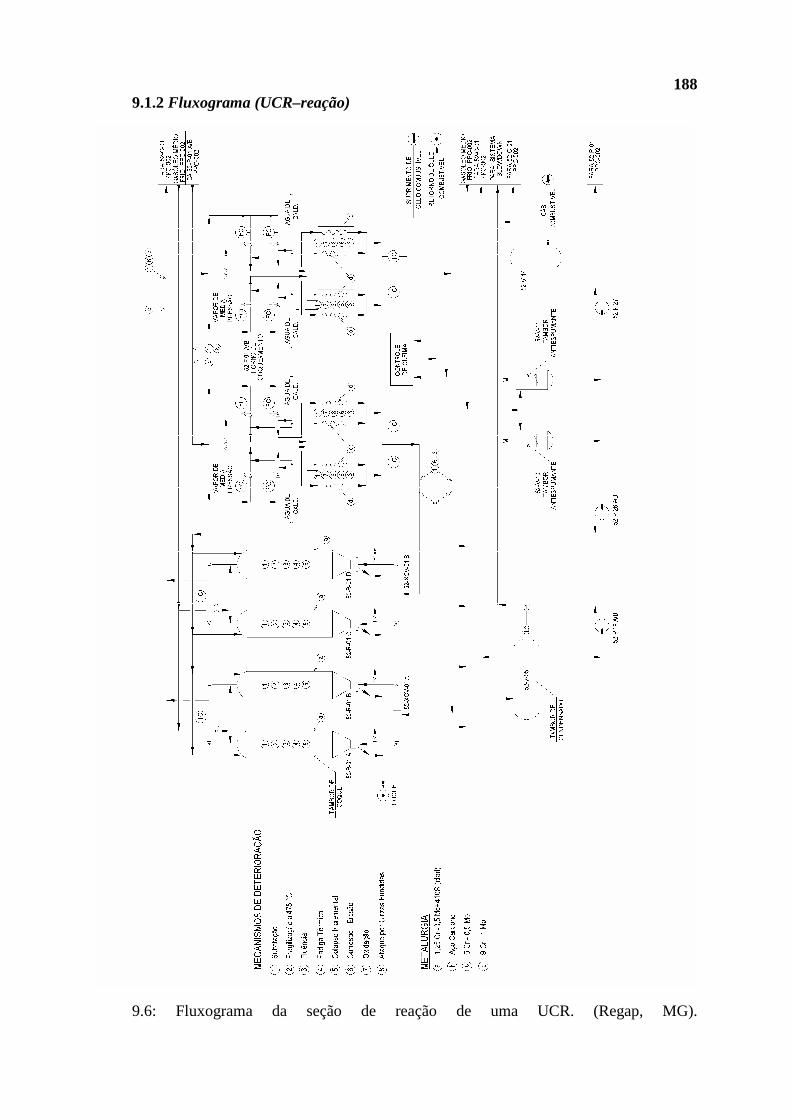
188 9.1.2 Fluxograma (UCR–reação)
9.6: Fluxograma da seção de reação de uma UCR. (Regap, MG).

189 CAPÍTULO 10
10.1 Conclusão
As ocorrências mais relevantes de corrosão e de deterioração do aço que se
desenvolvem em uma refinaria de petróleo que vem processando cargas ácisa, baixo
grau API, relativamente pobre em compostos sulfurados, foram discutidos nessa
dissertação. Em particular, foram considerados os processos que tendem a se
desenvolver nas destilações, a baixa e a alta temperatura, no craqueamento catalítico,
separação de gases e de tratamento DEA, hidrotratamento e geração de hidrogênio e por
fim o coqueamento retardado.
Em cada caso, foi procurado correlacionar o potencial que existe para o
desenvolvimento de um dado processo de corrosão e/ou fragilização em um
determinado equipamento ou trecho de uma planta de uma refinaria de petróleo. Para
tanto, empregou-se, na maior parte dos casos, informações originárias de casos reais de
corrosão ocorridos em uma refinaria. Tais dados, juntamente com a consulta à literatura,
terminaram por permitir fazer tais correlações e tecer considerações relativas à formas
de atenuá-los, apontando em fluxogramas de processo as posições em que são esperados
a ocorrência de danos específicos.

190 Referências Blibiográficas
1. BARBOSA, L. C. A. Introdução à Química Orgânica. Pearson/Prentice Hall, 2004
2. ATKINS, P. W. Moléculas. EDUSP, 2000
3. ABADIE, E. Processos de Refino. DIVEN, Petrobras, 1982.
4. SPEIGHT, J. G.; ÖZÜM B. Petroleum Refining Process, Marcel Dekker, 2002.
5. REIS, M. N. C. et alli. Curso de Formação de Operadores de Refinaria.
Unicenp/Petrobras-2002.
6. GÓMEZ, M. T. et alli. Formacion de Sedimentos Durante la Hidrodesintegracion de
Residuos del Petróleo. Revista de la Sociedad Química de México, vol. 47, n° 3,
2003.
7. SARAIVA, L. M. et alli. Avaliação dos Componentes do Resíduo de Destilação de
um Petróleo Pesado Brasileiro por Termogravimetria. V CBRATEC, 2006.
8. HELLE, H. P. E. Guideline for Corrosion Control in Crude Distillation Units. New
Plantation, 1994.
9. PAIVA, G. J. M. As Técnicas de Perda de Massa e de Resistência Elétrica na
Análise da Corrosão Naftênica em Laboratório e no Campo. Dissertação de
Mestrado, COPPE/UFRJ, 1999.
10. PHIEL, R. L. Naphthenic Acid Corrosion in Crude Distillation Units. MP, 1988.
11. HAUSLER, R. H.; COBLE, N. D. Corrosion Control in Crude Unit Overhead
Systems. Hydrocarbon Processing, 1972.
12. ASME VIII, Boiler & Pressure Vessel Code, div. 1-2, 2004.
13. Norma Petrobras, N-133, Soldagem, 2005.

191
14. ASME VIII-1, UCS-56, 2004.
15. BENHAYON, J. M. Introducción al Proyecto de las Construcciones Metálicas
Soldadas. Boletín Técnico Conarco, n°95, 1989.
16. DEVLETIAN, J. H.; WOOD, W. E. Principles of Joining Metalurgy. Metals
Handbook, ASM, Ninth Edition, vol. 6, 1983.
17. WAINER, E. et alli. Soldagem. Processos e Metalurgia. Ed. Edgard Blücher, 1992.
18. ALVISI P. P. Soldagem em Refinarias de Petróleo. Circulação Interna, Regap,
Petrobras, 2001.
19. EASTERLING K. Introduction to the Physical Metallurgy of Welding.
Butterworths, 1985.
20. Metals Handbook, ASM, vol. 11, 1986.
21. Norma Petrobrás, N-2301, Elaboração de Documentação Técnica de Soldagem,
2006.
22. FOLKHARD, E. et alli. Welding Metallurgy of Stainless Steels. Springer-Verlag
Wien New York, 1988.
23. MODENESI, P. J. Soldabilidade dos Aços Inoxidáveis. SENAI-ACESITA, 2001.
24. BAILEY, N. Welding Dissimilar Metals. The Welding Institute, 1986.
25. UHLIG, H. H. Corrosion y Control de Corrosion. Urmo, 1979.
26. TRETHEWEY, K. R.; CHAMBERLAIN, J. Corrosion for Students of Science and
Engineering. Longman Scientific&Technical, 1988.
27. GENTIL, V. Corrosão. LTC, 1996.

192
28. ALVISI, P. P. Furo em Tubo de Caldeira Flamotubular. Circulação Interna, Regap,
Petrobras, 2001.
29. WAARD, C.; LOTZ, U. Prediction of CO2 Corrosion of Carbon Steel.
Corrosion/93, paper 69, NACE, 1993.
30. ALVISI, P. P.; COUTO, V. S. Ataque Ácido em Torre Reneradora de Amina. 3º
Seminário Latino-Americano de Inspeção de Equipamentos, IBP, 1995.
31. ALVISI, P. P.; MORAES, D. A. R. Ataque Ácido nos Tubos da Zona de Convecção
de Caldeira do Sistema de Geração de Vapor da REGAP. Simpósio Nacional Sobre
Integridade em Centrais de Vapor, ABCM, 1991.
32. FONTANA, M. G.; GREENE, N. D. Corrosion Engineering. McGraw-Hill, 1982.
33. POURBAIX, M. Lições de Corrosão Eletroquímica. Ed. Instituto Nacional de
Investigação Industrial INII, Lisboa, 1987.
34. STANSBURY, E. E.; BUCHANAN, R. A. Fundamentals of Electrochemical
Corrosion. ASM, 2000.
35. DILLON, C. P. Corrosion of Steel by Caustic. MP, 1995.
36. KANE, R. D.; CAYARD, M. S. Understanding Critical factors that Influence
Refinery Crude Corrosiveness. MP, 1999.
37. NESÍC, S.; POSTLETHWAITE, J. Hydrodynamics of Disturbed Flow and Erosion-
Corrosion. Part I – Single Phase Flow Study. The Canadian Journal of Chemical
Engineering, vol. 69, 1991.
38. ALVISI, P. P.; SILVA, H. R. Caracterização da Ocorrência de Corrosão
Intergranular em um Aço AISI 405. 2º Seminário Latino-Americano de Inspeção de
Equipamentos, IBP, 1991.

193 39. TIMMINS, P. F. Predictive Corrosion and Failure Control in Process
Operations. ASM, 1996.
40. GAVELE, J. R. Past, Present, and Future of Stress Corrosion Cracking. Corrosion,
vol. 55, nº 8, NACE, 1999.
41. SEDRIKS, A. J. Corrosion of Stainless Steels. John Wiley&Sons, 1996.
42. LLEWELLYN, D. T. Steels: Metallurgy and Applications. Butterworth Heinemann,
1994.
43. TELLES, P. C. S. Materiais para Equipamentos de Processo. Ed. Interciência,
2003.
44. DIETER, G. E. Mechanical Metallurgy. McGraw-Hill, 1976.
45. SMALLMAN, R. E. Modern Physical Metallurgy. Butterworth, 1985.
46. BRAGA, J. A.; ALVISI, P. P. Liga SB-443. Uma Avaliação à Susceptibilidade ao
Ataque Intergranular, 2º Seminário Latino-Americano de Inspeção de
Equipamentos, IBP, 1991.
47. API RP-530. Calculation of Heater-Tube Thickness in Petroleum Refineries. 2001.
48. HERTZBERG, R. W. Deformation and Fracture Mechanics of Engineering
Materials. John Willey&Sons, 1989.
49. SILVEIRA, T. L.; MONTEIRO, S. N. Controle da Vida Remanescente em
Equipamentos Industriais que Operam em Condições de Fluência. ABM, vol. 37, nº
281, 1981.
50. ALVISI, P. P. Carbonetação Localizada e Pontos de Sobre-Aquecimento em
Colunas de Reforma HK-40. 19º Seminário de Inspeção de Equipamentos, IBP,
1992.

194 51. SILVEIRA, T. L. et alli. Análise de Integridade em Equipamentos Estáticos que
Operam em Altas Temperaturas na Indústria Petroquímica e no Refino do Petróleo.
3º Colóquio Latino-Americano de Fadiga e Fratura dos Metais, 1989.
52. ALVISI, P. P. Ruptura Prematura em Tubo de Forno de Reforma a Vapor por
Fluência Assistida por Carbonetação Severa. Circulação Interna, Regap, Petrobras,
1996.
53. LAI, G. Y. High-Temperature Corrosion of Engineering Alloys. ASM
International,1997.
54. BEDNAR, H. H. Pressure Vessel Design Handbook. Krieger Publishing Co., 1991.
55. PIEPER, C. J. et alli. Coke Drum Design-Longer Life Through Innovation. AIChE,
2000.
56. PENSO, J. A. et alli. Assessing Deterioration Conditions in Coke Drums. Welding
Journal, 2000.
57. STEWART, C. Vertical Plate Technology Extend the Life of Coke Drums. Welding
Journal, 2004.
58. MELLO, T. L. Esferoidização e Grafitização em Aços de Componentes
Petroquímicos. Dissertação de Mestrado, UFRS, 1983.
59. BIRKS, N.; MEIER, G. H. Introduction to High Temperature Oxidation of Metals.
Edward Arnold, 1983.
60. GASKELL, D. R. Introduction to Metallurgical Thermodynamics. McGraw-Hill
Co., 1977.
61. SHIGLEY, J. E. Mechanical Engineering Design. McGraw-Hill, 1986.
62. JASTRZEBSKI, Z. D. The Nature and Properties of Engineering Materials. John
Wiley & Sons, 1977.

195 63. WULFF, J. et alli. Ciências dos Materiais. Livros Técnicos e Científicos Ed.,
vol. 2, 1972.
64. ALVISI, P. P.; SILVA, H. R. Ocorrência de Carbonetação e Oxidação na Câmara
Plena de um Regenerador de uma Unidade de Craqueamento Catalítico. 16º
Seminário de Inspeção de Equipamentos, IBP, 1988.
65. GRAKBE, H. J. Carburization. A High Temperature Corrosion Phenomenon. MTI
Pubications, Nº 52, 1998.
66. FORSETH, S.; KOFSTAD, P. Metal Dusting Phenomenon During Carburization of
FeCrNi-alloys at 850-1000 ºC. Materials and Corrosion, 46, 1995.
67. API Publ. 581. Risk-Based Inspection. 2000.
68. API RP 941. Steels for Hydrogen Service at Elevated Temperatures and Pressures
in Petroleum Refineries and Petrochemical Plants. 2004.
69. TIMMINS, P. F. Solutions to Hydrogen Attack in Steels, ASM, 1997
70. Norma Petrobras, N-1704, Projeto de Vaso de Pressão para Serviço com H2, rev. B,
2002.
71. ANTALFFY, L. P.; CHAKU, P. N. US Perspective of Modern Hydroprocessing
Reactor Metallurgy. HTI Quarterly, 1996.
72. ALVISI, P. P. Aços para Reatores HDT. Circulação Interna, Regap, Petrobras,
2006.
73. API RP 934. Materials and Fabrication Requirements for 2,25Cr-1Mo & 3Cr-1Mo
Steel Heavy Wall Pressure Vessels for High Temperature. High Pressure Hydrogen
Service, 2000.
74. Technical Specification, Additional Requirements for Cr-Mo Low Alloy Steel,
Cenpes, Petrobras, 2005.

196
75. PECKNER, D.; BERNSTEIN, I. M. Handbook of Stainless Steels, 1977.
76. NACE Standard RP0170-92. Protection of Austenitic Stainless Steels and Other
Austenitic Alloys from Polythionic Acid Stress Corrosion Cracking During
Shutdown of Refinery Equipment. NACE, 1997.
77. WU, Y. M. Calculations Estimate Process Stream Depositions. Oil&Gas Journal,
1994.
78. HARVEY, C.; SINGH, A. Mitigate Failures for Reactor Effluent Air Coolers,
Hydrocarbon Processing, 1999.
79. ALVISI, P. P.; ECKSTEIN, C. B. Nota Sobre a Ocorrência de Sal Ácido na U-210.
Circulação Interna, Regap, Petrobras, 2006.
80. SLAVCHEVA, E. et alli. Review of Naphthenic Acid Corrosion in Oil Refining.
British Corrosion Journal, vol. 34, 1999.
81. TEBBAL, S.; KANE, R. D. Critical Factors Affecting Crude Corrosivity. PTQ
Spring, 1997.
82. KIBALA, E. B. et alli. Special Report Section: Identify and Prevent Refinery
Naphthenic Acid Corrosion, FUEL, 1994.
83. PIEHL, R. L. Naphthenic Acid Corrosion in Crude Distillation Units. MP, vol. 27,
1988.
84. NUGENT, M. J.; DOBIS, J. D. Experience with Naphethenic Acid Corrosion in
Low TAN Crudes. Corrosion98, paper 577, 1998.
85. ALVISI, P. P.; COUTO, V. S. Nota Sobre a Ocorrência de Corrosão Naftênica em
uma Unidade de Destilação a Vácuo. Circulação Interna, Regap, Petrobras, 2003.
86. MATHERS, R. Treatment of High Acid Crudes and the Methods Used in Refineries
to Mitigate Naphthenic Acid Corrosion, EUROCORR/2005, 2005.

197
87. AMEER, M. A. et alli. Electrochemical Behavior of Passive Films on Molybdenum-
Contaninig Austenitic Stainless Steels in Aqueous Solution. Electrochimica Acta,
2004.
88. TURNBULL, A. et alli. Factors Controlling Naphthenic Acid Corrosion. NACE,
1998.
89. BERNARDES, R. Comportamento de Aços Inoxidáveis Comerciais e Inconel na
Resistência a Corrosão Naftênica de Petróleos Nacionais: Uma Análise
Fenomenológica. Dissertação de Mestrado, COPPE, UFRJ, 2005.
90. EFIRD, K. D. Jet Impingement Testing for Flow Accelerated Corrosion. Corrosion-
2000, paper 52, NACE, 2000.
91. LEWIS, K. R. et alli. Processing Corrosive Crude Oils. Corrosion-99, paper nº 377,
NACE, 1999.
92. ALVISI, P. P. Ocorrência de Corrosão Naftênica em Carcaça de Bomba de Diesel
Leve. XXII CONBRASCORR, 2002.
93. KAESCHE, H. Metallic Corrosion, NACE, 1985.
94. CHRISTENSEN, C. et alli. Hydrogen Permeation/Concentration Effects in Line
Pipe and Pressure Vessel Steels as a Result of Wet Sour Corrosion. Corrosion92,
paper nº 40, NACE, 1992.
95. AUBERT, L. Contribuition of Mechanism in the Modelling of Environment
Sensitive Fracture, EUROCORR, 2005.
96. WILHELM, M.; ABAYARATHNA, D. Inhibition of Absorption of Hydrogen by
Steels in Wet H2S Refinery Environments. Corrosion92, paper n° 449, NACE,
1992.

198 97. IYER, R. N. rt alli. Hydrogen Sulfide Effect on Hydrogen Entry into Iron-A
Mechanistic Study. Corrosion, NACE, 1990.
98. BONNER, W. A.; BURNHAM, H. D. Air Injection for Prevention of Hydrogen
Penetration of Steel. NACE, 1955.
99. EHMKE, E. F. Polysulfide Stops FCCU Corrosion. Hydrocarbon Processing, 1981.
100. VAN GELDER, K. et alli. Approach to Determine the Fiftness for Service of
Pipeline Steels with Respect to Hydrogen-Related Cracking. MPC Proceedings,
API, 1989.
101. ALVISI, P. P. Efeito da Concentração de Cianetos na Nucleação de HIC’s.
Circulação Interna, Regap, Petrobras, 2003.
102. ALVISI, P. P. Apresentação de um Critério para a Avaliação e o
Acompanhamento de Fissuras Induzidas pelo Hidrogênio com Desenvolvimento
em Degrau. 20° Seminário de Inspeção de Equipamentos, 2001.
103. MERRICK, R. D. Refinary Experiences with Cracking in Wet H2S
Environments. MP,1988.
104. Norma Petrobras, N-1706, Projeto de Vaso de Pressão para Serviço com H2S,
rev. C, 2004.
105. KANE, R. D. et alli. An Exploratory Examination of the Effect of SOHIC
Damage on the Fracture Resistence of Carbon Steels. Corrsionsource/2000,
paper 1101, 2000.
106. MORIN, C. R. et alli. Analysis of Causative Factors of the MEA Absorber
Vessel Rupture. Chicago, Illinois, July 23, 1984, Failure Analysis submitted to
the Illinois Office of the State Fire Marshall, Parker Engineering Associates Inc.,
1985.

199 107. NACE MR 0175 part 2, Cracking–Resistent Carbon and Low Steels, annex
A, A2.1.4, 2003.
108. ALVISI, P. P.; COELHO, P. C. Limitando em 22HRC a Dureza de uma Junta
Soldada em Aço A515-70 Através da Especificação da Energia de Soldagem. I
ENSOLD, 2004.
109. EATON, P. et alli. The Impact of Napthenic on Salt Hydrolysis. Eurocorr,
Lisboa, 2005.
110. PETERSEN, R. P. et alli. Choosing a Neutralising Amine Corrosion Inhibitor.
PTQ Summer, 2004.
111. ALVISI, P. P. Otimização da Injeção de Soda na Destilação. Circulação Interna,
Regap, Petrobrás, 2005.
112. ALVISI, P. P. Processos Corrosivos Associados à Depósitos. Sistema de Topo
da Destilação Atmosférica. 25ºSEMINSP, 1999.
113. ALVISI, P. P.; GOMES, M. S. Ocorrência de Trincamento Intergranular por
CST por Carbonato em Linha de Topo de Torre Fracionadora UFCC.
Circulação Interna, Regap, Petrobras, 2004.
114. DUPART, M. S. et alli. Understanding Corrosion in Alkanolamine Gas Treating
Plants. Parts ½. Hydrocarbon Processing, 1993.
115. HAWS, R. Contaminants in Amine Gas Treatment. GPA Houston Regional
Meeting, 2001.
116. CORREA, L. A. et alli. Corrosion Control by On-Line Monitoring in Gas
Treatment Units. 2nd NACE Latin American Region Corrosion, 1996.
117. API RP 945, Avoiding Environmental Cracking in Amine Units, 1997.
118. HAKKA, L. E. et alli. The Role of Anion Contaminants on Corrosion in Refinery
Amine Units. Gas Treating Products&Sevices, DOW, 1995.

200
119. SHAO, J. et alli. Removal of Heat Stable Salts – A Solution to Amine Plant
Operational Problems. SM/Technical Paper 157, 2002.
120. Instrução Operacional/U-210, Regap, Petrobras,2004.
121. ALVISI, P. P. Ruptura Prematura de Tubo de Forno de Reforma a Vapor por
Fluência Assistida por Carbonetação Severa. Circulação Interna, Regap,
Petrobras, 1996.
122. JASKE, C. S. et alli. Predict Reformer Furnace Tube Life. Hydrocarbon
Processing, 1983.
123. RIBEIRO, E. A. A. et alli. 25Cr-35Ni-Nb+Microadditions Alloys. A Reliable
Choice for Petrochemical Industries. Villares, Steel Division, 1993.
124. ALVISI, P. P. et alli. Possibilidades de falha por Fluência dos Componentes do
″Manifold″ de Saída do 209-F-02. Circulação Interna, Regap, Petrobras, 2004.
125. THOMAS, C. W.; KNOWLES, D. M. Embrittlemente in Cast Reformer Outlet
Manifold Components. Ammonia Technical Manual, 2003.
126. MONTEIRO, S. N. High-Temperature Failure by Perforation of Incoloy 800H
Pigtails in Reformer Furnaces. Corrosion in the Petrochemical Industry, ASM,
1994.
127. ALVISI, P. P. Nota Sobre o Sistema Coletor do Forno de Reforma 111-F-02.
Circulação Interna, Regap, Petrobras, 2001.
128. ALVISI, P. P. Ataque Ácido e Falha do 209-E-04. Circulação Interna, Regap,
Petrobras, 2005.
129. Módulo de Treinamento, DIPRO/SECOQ, REGAP, PETROBRAS, 2004.

201 130. ALVISI, P. P. Efeitos do Coque nos Tubos dos Fornos de Processo. Anexo
3, Relatório de Análise de Falha. Convecção do 52-F-01B, 2004, Circulação
Interna, Regap, Petrobras, 2004.


















